Introduction
It has been estimated that around 285 million people suffer from type 2 diabetes mellitus (T2DM) with projected rise to 438 million in the next 20 years (1). Current therapies for T2DM include lifestyle modification and use of oral antidiabetic drugs (OAD). Several OAD classes are currently available to treat T2DM patients, with sulphonylureas (SU), biguanides, thiazolidinediones (TZDs), and meglitnides being the most frequently used. Effects of these drugs depend on the extent of drug absorption from the gut lumen, uptake and metabolism of the drug in the liver, the extent of its transport back into the systemic circulation for extrahepatic effects, and for drugs with substantial renal secretory clearance (e.g., metformin) on active renal tubular secretion into the urine. Drug-metabolizing enzymes (DME) and drug transporters (DT) have a crucial role in the fate of drugs in human body. Information regarding the incidence of cytochrome P450 (CYP) gene polymorphisms is increasing (2-4), although the level of detail such as in the case of other DME, including UDP-glucuronosyltransferases and N-acetyltransferases, is not as extensive. In addition, recent studies identified genetic variations of DT associated with the altered treatment outcomes (5,6) and revealed an important role of the transporter-mediated OAD uptake in their disposition and effects (4).
Furthermore, recent data showed that epigenetic changes in response to environmental stimuli may play an important role in the diabetes development (7). In addition to the role of DNA methylation and histone modifications, recent studies also indicated that microRNAs (miRNAs), a class of small, single-stranded non-protein coding gene products that post-transcriptionally suppress mRNA target expression, could serve as a new generation of biomarkers for diabetes. Strikingly, a recent analysis of the specific miRNAs provided sufficient data to diagnose and predict the T2DM development in normoglycemic patients (8). Thus, characterization of the miRNAs pattern together with the analysis of specific pharmacogenetic variants could facilitate the design of a therapy tailored to the individual patient, contributing to the current efforts in the personalized medicine (9,10). Here we aim to summarize the pharmacogenomics data related to the T2DM therapy, particularly the associations of SU, TZDs, biguanides, and meglitinides treatment outcomes with polymorphisms of DME, DT, drug target and diabetes risk genes that might lead to the promotion of personalized T2DM treatment.
Sulphonylureas
In addition to insulin resistance (IR), insufficient pancreatic β-cell function plays an important role in the pathogenesis of T2DM. The insufficient production and secretion of insulin can be increased by secretagogue drugs, like SU that bind to sulphonylurea receptor (SUR) leading to the cell depolarization and stimulation of insulin release. Potassium inwardly rectifier 6.2 subunit (Kir6.2) of pancreatic islet ATP-sensitive K+ (KATP) channels are heterooctamers assembled from Kir6.2 and the sulphonylurea receptor 1 (SUR1), encoded by the KCNJ11and ABCC8 gene, respectively. This channel is essential for glucose-stimulated insulin secretion from pancreatic β-cells, modulates glucose uptake into skeletal muscle, glucose production and release from the liver (11). Physiologically, decreased plasma glucose levels lead to lower metabolic rate which opens KATP channels, suppressing electrical activity and insulin release. In contrast, KATP channels close when metabolism increases, ATP rises and MgADP falls, leading to membrane depolarization, opening of voltage-gated Ca2+ channels, Ca2+ influx, and insulin secretion.
In recent years single nucleotide polymorphisms (SNPs) of the genes encoding KATP channel have been related to the efficacy of secretagogue drugs (Table 1). Loss-of-function (LOF) mutations of these genes are the most common cause of congenital hyperinsulinism, with over 150 mutations characterized in SUR1 (ABCC8) and 24 in Kir6.2 (KCNJ11) (12). On the other hand, gain-of-function (GOF) mutations in Kir6.2 or SUR1 cause neonatal diabetes (ND). A similar number of over 40 different ND mutations have been characterized in Kir6.2 and in SUR1 (12). All Kir6.2 mutations cause dominant ND by reducing the ability of ATP to block KATP channel (13), hyperpolarizing the β-cells, and preventing insulin secretion. Thus, the mutated KATP channel does not close in response to increased ATP concentrations. However, it could be closed when SU bind to SUR1 by an ATP-independent route. The discovery of the causal role of KATP channels has enabled ND patients to switch from insulin to SU therapy that significantly improved glycemic control and reduced the risk of diabetic complications (14,15).
Table 1. Summary of the gene polymorphisms involved in the pharmacogenetics of sulphonylureas.
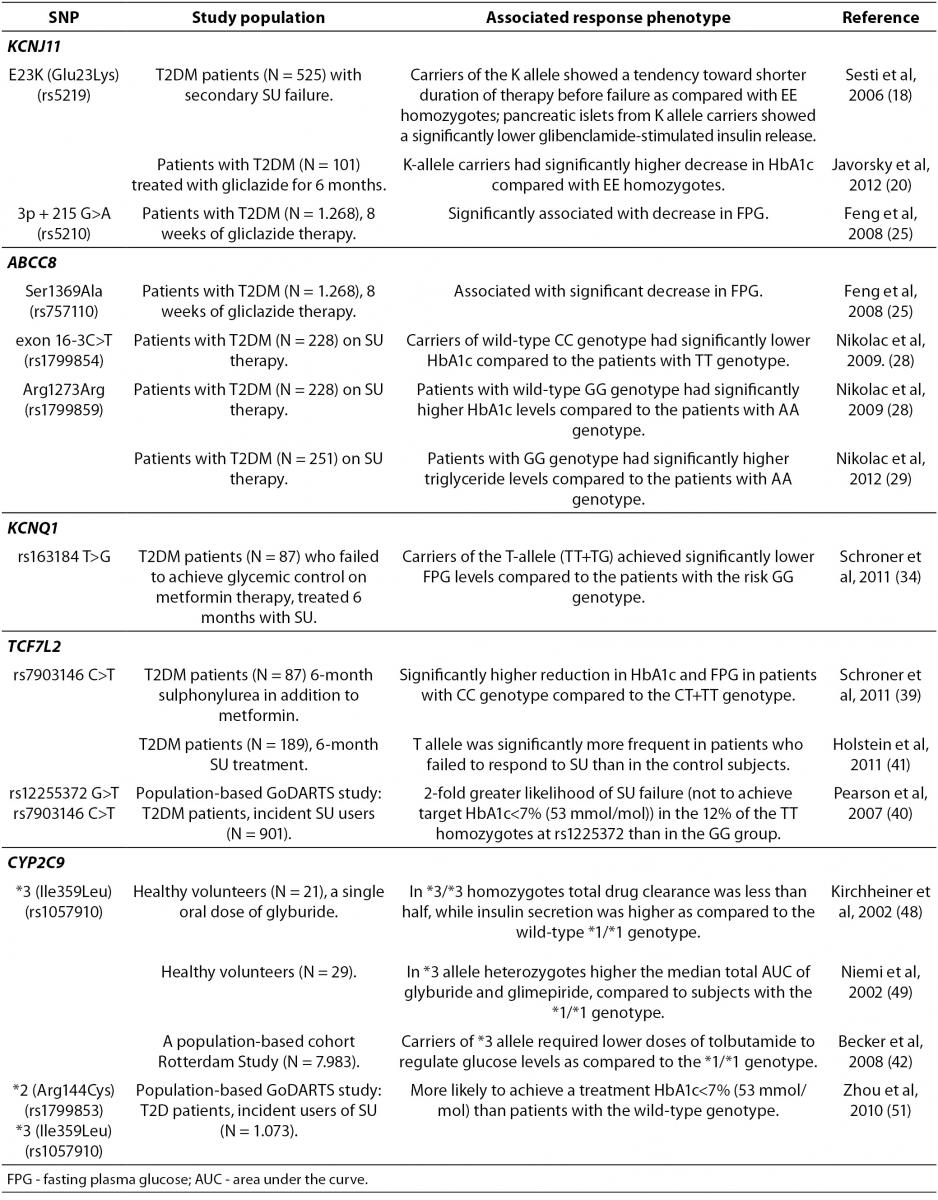
A common Glu23Lys polymorphism (also known as E23K) in KCNJ11 (potassium inwardly-rectifying channel, subfamily J, member 11) gene encoding Kir6.2, is associated with T2DM development (16,17), as well as an increased risk of SU therapeutic failure (18). The functional effects of the E23K variant on insulin secretion and insulin sensitivity in humans are controversial, although recent larger studies demonstrate a significantly reduced insulin secretion, lower insulin levels, and improved insulin sensitivity (19), consistent with enhanced KATP activity in pancreatic β-cells. Furthermore, a recent study found that the carriers of the KCNJ11 K-allele had better therapeutic response to gliclazide (20). KCNJ11 variations have been also associated with altered response to glibenclamide therapy and misdiagnosis of Type 1 diabetes (14). Importantly, recent evidence demonstrated that patients with KCNJ11 mutations could be treated more efficiently with SU than with insulin (14,21,22).
The ABCC8 gene encodes the SUR1 subunit which regulates KATP channel activity. ABCC8 mutations are genetically more heterogeneous, with homozygous, heterozygous and compound heterozygous mutations being described (23). Heterozygous activating mutations in the ABCC8 gene have been characterized as a cause of permanent and transient ND that may present as T2DM (24). Interestingly, a common Ser1369Ala SNP of ABCC8 influenced antidiabetic efficacy of SU in Chinese (25), but not in German population (26). In addition, this same Ser1369Ala variant of ABCC8 appeared not to be associated with the risk for severe SU-induced hypoglycemia in German (26) and Japanese (27) T2DM patients. Additional polymorphisms of ABCC8 gene, including SNP in exon 16 (-3C/T) and exon 31 (Arg1273Arg) have been also reported to be associated with the SU efficacy in European Caucasians (28-30).
Importantly, two common ATP-sensitive potassium (KATP) channel variants, E23K and S1369A, of the KCNJ11 and ABCC8 genes respectively, are in strong linkage disequilibrium (LD) and form a haplotype that appears to be associated with an increased T2DM risk (31). A recent analysis of structure-activity relationships in KATP channels containing either the E23/S1369 non-risk or K23/A1369 risk haplotypes, demonstrated that KATP channels containing the K23/A1369 risk haplotype were significantly less sensitive to inhibition by tolbutamide, chlorpropamide, and glimepiride (32). Glibenclamide and glipizide demonstrated similar inhibitory effects on KATP channels in patients with either haplotype. Based on these data, the authors suggested that it would be possible to design novel OAD with an increased efficacy in patients homozygous for these common KATP channel haplotypes.
Patients with deficient hepatic nuclear factor-1-alpha (HNF1-alpha), a transcription factor vital for correct β-cell development and function, have progressive β-cell deterioration and are more sensitive to SU than matched T2DM patients (33). Furthermore, a recent study has suggested that the magnitude of fasting plasma glucose (FPG) levels reduction after 6-month SU treatment in addition to metformin in T2DM patients was related to the rs163184 (T>G) SNP in KCNQ1 gene (34). KCNQ1 encodes a voltage-gated potassium channel expressed in the heart, stomach, small and large intestine, kidney, and pancreas. However, its role in insulin secretion by pancreatic β-cells is not completely understood. Importantly, the FPG response to SU was significantly lower in carriers of the risk GG genotype of rs163184 variation in KCNQ1(34). In addition, the Arg(972) variant of IRS-1 gene encoding the insulin receptor substrate (IRS)-1 that is an important component of the insulin signaling cascade, was also associated with an increased risk for secondary SU failure (35).
The most promising gene variants affecting the SU response are those involved in drug pharmacodynamics, such as the transcription factor 7-like 2 (TCF7L2) that encodes a transcription factor (Tcf-4), involved in the regulation of cellular proliferation and differentiation (36). TCF7L2gene has the strongest association with T2DM reported to date (37,38). A recent study demonstrated that the degree of reduction in HbA1c and FPG levels following a combined SU and metformin treatment was related to TCF7L2 rs7903146 (C>T) SNP (39). Interestingly, individuals with the T2DM-associated homozygous TT genotype were less likely to respond to SU therapy (39-41).
Sulphonylureas are mainly metabolized by the enzyme cytochrome P450 (CYP) isoform CYP2C9. A recent population-based Rotterdam study showed that polymorphisms in CYP2C9 gene affected the patient sensitivity to SU (42). The carriers of CYP2C9*3 (Ile359Leu) polymorphism appeared to be protected against development of T2DM (43), while CYP2C9*2 (Arg144Cys) polymorphism was not associated with diabetes susceptibility (44). Although these two allelic variants, CYP2C9*2 and CYP2C9*3, have been associated with elevated SU serum levels, the tolbutamide dose was lower in CYP2C9*3 allele carriers as compared to patients with the wild-type CYP2C9 genotype, while similar effect was not observed for the CYP2C9*2 genotype (42). Furthermore, CYP2C9*3 increased the risk of hypoglycemia in T2DM patients treated with SU (43,45) and multiple studies indicated that the CYP2C9*3 allele was associated with decreased SU clearance (46,47). Homozygous carriers of the CYP2C9*3/*3 genotypes had reduced clearance of glyburide and increased insulin secretion following glyburide administration (48, 49). Furthermore, diabetic patients with a CYP2C9*3 variant required lower doses of tolbutamide for glucose control than patients with the wild-type genotype (42). Recently, Swen et al. (50) demonstrated no significant effects of the CYP2C9*2 and CYP2C9*3 alleles on prescribed dose in incident SU users. However, a large population-based GoDARTS (Genetics of Diabetes Audit and Research in Tayside Scotland) study performed in 1,073 incident SU users demonstrated that patients with two copies of the *2 or *3 alleles were three times more likely to achieve treatment target of HbA1C levels under 7% (53 mmol/mol) than patients with two wild-type CYP2C9*1alleles (51).
Thiazolidinediones
Thiazolidinediones,also known as glitazones, act by activating their molecular target, PPARs (peroxisome proliferator-activated receptors). TZDs bind with greatest specificity for PPARγ, a nuclear transcription factor expressed diffusely in humans, including adipocytes, to promote adipogenesis and fatty acid uptake. By reducing circulating fatty acid concentrations and lipid availability in liver and muscle, these drugs improve the patients’ sensitivity to insulin and reduce hyperglycemia. In addition, TZDs have a multitude of other therapeutic effects including anti-inflammatory effects and amelioration of hypertension, microalbuminuria and hepatic steatosis (52).
Rosiglitazone and pioglitazone are TZDs that have been shown to improve glycemic control and may act to slow the progression of β-cell failure. However, recent studies demonstrated that use of rosiglitazone could cause serious side effects, including an increased risk of myocardial infarction and death from cardiovascular causes as compared to pioglitazone (53,54). Due to these serious side effects, the European Medicines Agency (EMA) has recommended the suspension of the marketing authorizations for the rosiglitazone, while US Food and Drug Administration (FDA) has decided that rosiglitazone can remain available, but only under a very stringent restricted access program. Thus, the only TZD that is still available on market is pioglitazone, which appears to have more favorable effects on cardiovascular system in T2DM patients as compared to rosiglitazone (55,56). The mechanism of the antidiabetic action of pioglitazone involves activation of PPARγ, while its direct antioxidant action may also contribute to its effect on IR (57). However, the treatment with pioglitazone for more than one year may be associated with an increased risk of bladder cancer (58-60).
Recently, as shown in Table 2, several gene variants have been also associated with the TZDs therapy outcomes (61). Adipokinins, including adiponectin, leptin, resistin, peroxisome proliferator-activated receptor γ (PPARγ) and tumor necrosis factor (TNF)-α, are of a particular interest due to their important role in IR. Loss-of-function mutations in PPARγ are associated with severe IR and diabetes mellitus (62). Variants in PPARG or ADIPOQ (adiponectin) have been variably associated with TZDs response. Strikingly, rosiglitazone was significantly more effective in diabetic patients with Pro12Ala polymorphism of PPARG, who consequently had a significantly greater decrease in FPG and HbA1c levels than carriers of the wild-type genotype (63). A recent study reported that Thr394Thr and Gly482Ser SNPs of the peroxisome proliferator activated receptor-γ coactivator-1α (PGC-1α), which is a transcriptional coactivator of PPARγ, were also associated with rosiglitazone efficacy in Chinese T2DM patients (64). In addition, a pilot study suggested that the G/G genotype of resistin SNP-420 may be an independent predictor of the reduction of FPG and HOMA-IR (Homeostasis Model of Assessment-Insulin Resistance) by pioglitazone (65). Furthermore, recent studies demonstrated that the genetic variations of –11377C/G and T45G in adiponectin gene (66) and leptin G-2548A (67), as well as TNF-α G-308A (67) appeared to affect rosiglitazone efficacy and reversed IR in Chinese diabetic patients.
Table 2. Summary of the gene polymorphisms involved in the pharmacogenetics of thiazolidinediones.
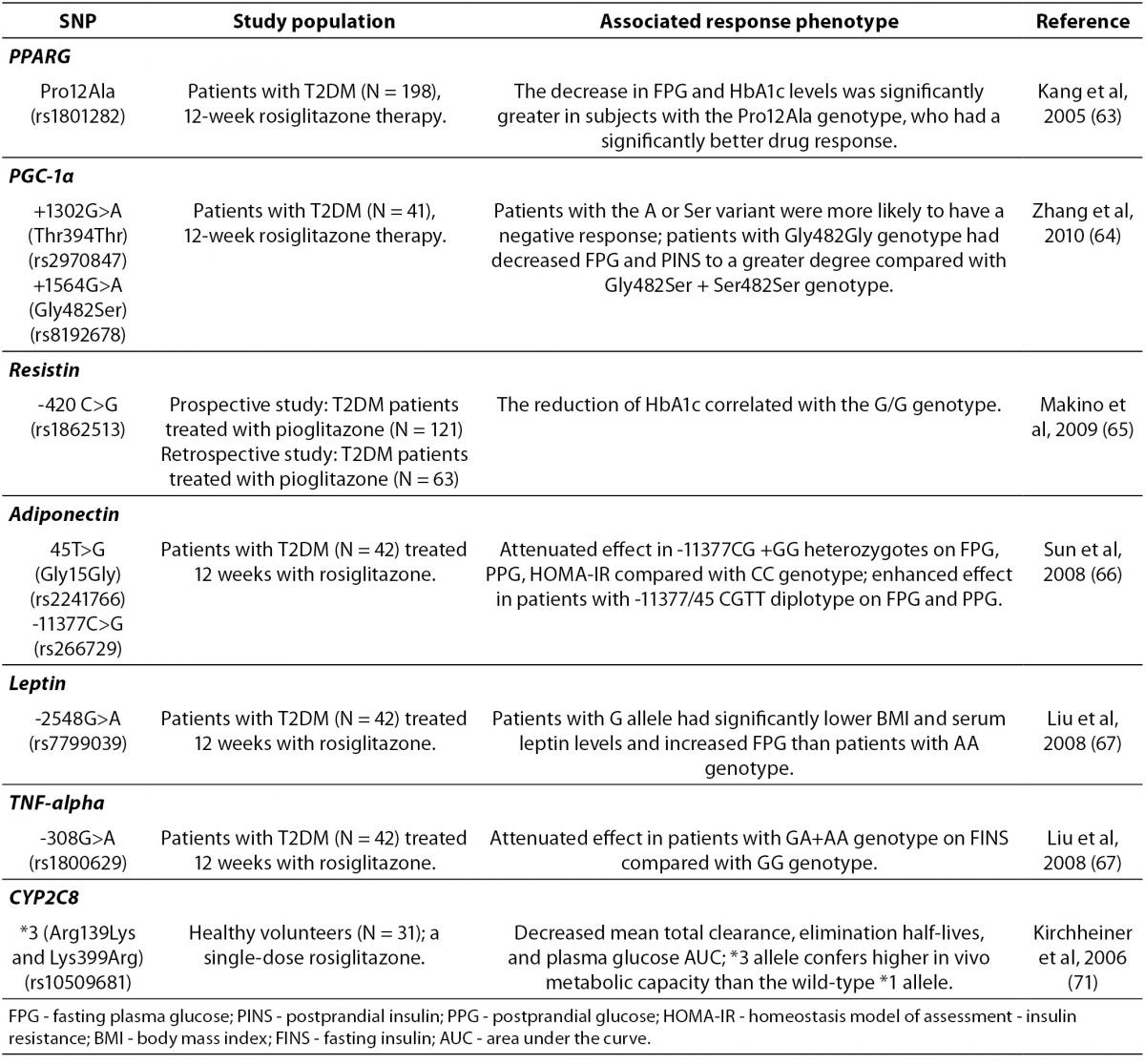
Treatment with TZDs has been particularly associated with high rates of developing fluid retention and peripheral edema, leading to congestive heart failure (52). Interestingly, a recent study done by Chang et al. (68) have suggested that rs296766 polymorphism of AQP2 gene codingaquaporin-2 (the vasopressin-regulated water channel), and rs12904216 polymorphism of SLC12A1 (the solute carrier protein family 12 group A, member one) gene coding the sodium-potassium-2 chloride transporter (NKCC2) with a key role in electrolyte movement across epithelia, represented risk factors for TZDs-associated edema. In addition to these genetic factors, the same study demonstrated that female gender and older age were also contributing factors to the edema development following TZDs treatment.
A recent study showed a diminished effect of pioglitazone treatment on glycemic control in patients with Ser447X polymorphism of gene encoding lipoprotein lipase (LPL), the enzyme responsible for the lipolytic processing of triglyceride-rich lipoproteins (61). CYP2C8 and CYP3A4 are the main DME catalyzing biotransformation of the pioglitazone (69), whereas rosiglitazone is metabolized by CYP2C8 and CYP2C9 (70). Genetic variation in CYP2C8 was found to be associated with altered clearance of rosiglitazone, with higher drug clearance in carriers of CYP2C8*3 allele coding for Arg139Lys and Lys399Arg amino acid substitutions (71).
Biguanides
The biguanide drug metformin is recommended as the first-line therapy for T2DM (72). A main action of metformin is suppression of hepatic gluconeogenesis (73). Other antihyperglycemic effects include an increase of glucose uptake and utilization, improvement of insulin sensitivity, and reduction of intestinal glucose absorption (73). The molecular mechanisms of metformin action are not fully elucidated. It is believed that metformin increases the AMP/ATP ratio by specific inhibition of mitochondrial respiratory-chain complex 1, which leads to the activation of adenosine monophosphate-activated protein kinase (AMPK) (74-77). Potential mechanism of AMPK phosphorylation by metformin includes the upstream serine-threonine kinase11 (STK11/LKB1) (78). However, a recent study showed that metformin could also inhibit gluconeogenesis in an AMPK-independent way (79).
Metformin is not metabolized but is excreted unchanged in urine by active tubular secretion (80). There is large variation in metformin renal clearance, and genetic factors contribute to this by more than 90% (81,82). Its oral absorption, hepatic uptake and renal elimination are mediated by carrier proteins (83). The intestinal absorption of metformin is probably mediated by plasma membrane monoamine transporter (PMAT, encoded by SLC29A4 gene), expressed on the luminal side of enterocytes (84). Organic cation transporter 1 (OCT1, encoded by SLC22A1 gene), expressed in the basolateral membrane of hepatocytes, mediates hepatic metformin uptake (85). OCT2 (encoded by SLC22A2), expressed primarily at the basolateral membrane in the kidney tubular cells, facilitates uptake of metformin into proximal tubule cells (86). The multidrug and toxin extrusion transporter 1 (MATE1, encoded by SLC47A1) and MATE2-K (encoded by SLC47A2), located in the apical membrane of the renal proximal tubule cells, facilitate metformin excretion from tubular cells into urine (87-89).
The therapeutic response to metformin is highly variable. In patients receiving metformin as an initial treatment for T2DM, less than two-thirds achieve desired glycemic control or the HbA1c goal of < 7% (< 53 mmol/mol) (90). This implies that identification of genetic factors associated with treatment effectiveness could be relevant. The majority of pharmacogenetic studies of metformin response have investigated the effects of polymorphisms in the gene encoding OCT1, SLC22A1 (Table 3). OCT1 is necessary for metformin transport into the liver and subsequent metformin activity. Human SLC22A1 gene is highly polymorphic. Reduced-function polymorphisms of the SLC22A1 gene, such as R61C (rs12208357), G401S (rs34130495), 420del (rs72552763), and G465R (rs34059508) have been associated with lower effects of metformin in the oral glucose tolerance test (91). These same SNPs were also associated with a significantly higher metformin AUC, higher maximal plasma concentration and lower oral volume of distribution (92). However, a subsequent study demonstrated an additive increase in renal clearance of metformin with increasing number of reduced-function alleles (93). In a large population-based GoDARTS study the effects of R61C and 420del variants on initial glycemic response to metformin, the mid-term HbA1c control, and the rate of metformin monotherapy failure were not found (94). Another study analyzed the effects of 11 tagging SNPs in the SLC22A1 gene on HbA1c reduction, in a smaller cohort of incident users of metformin (95). A significant association of intronic rs622342 A>C SNP and lower HbA1c reduction was found (95).
Table 3. Summary of the gene polymorphisms involved in the pharmacogenetics of biguanides.
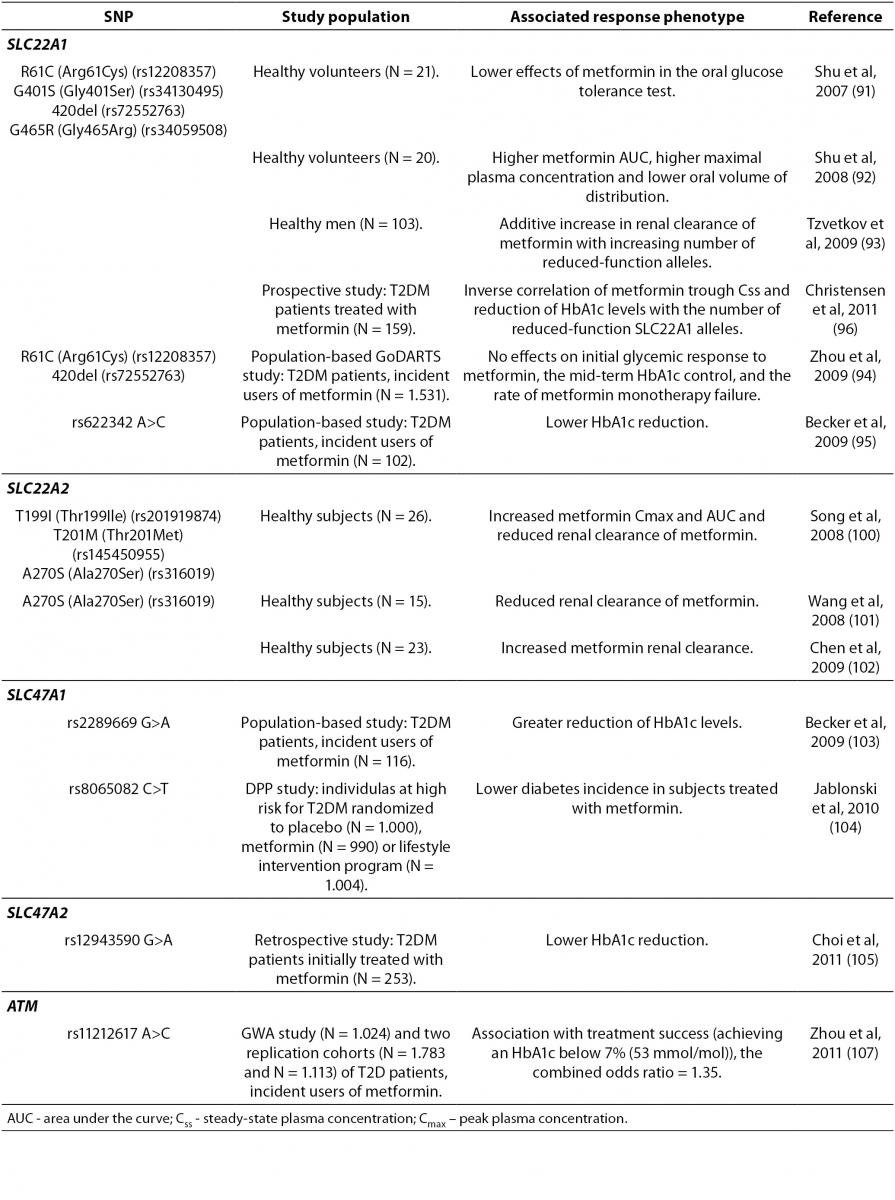
A recent prospective study investigated the effects of variations in genes encoding OCT1, OCT2, MATE1, MATE2, and PMAT, on the trough steady-state plasma concentration (Css) of metformin and HbA1c change in T2DM patients treated with metformin (96). The mean trough Css of metformin, as well as the absolute decrease in HbA1c levels after 6 and 24 months, correlated conversely with the number of reduced-function SLC22A1 alleles (R61C, G401S, M420del and G465R) (96). It is hypothesized that in patients with reduced function OCT1 variants, less metformin is transported into hepatocytes and the volume of distribution decreases, resulting in a shorter half-life and lower trough Css of metformin (96). These results are in accordance with previous studies (91,93). Replication of results regarding influence of OCT1 low-activity alleles on metformin pharmacokinetics and response, indicated that those variants could be useful pharmacogenetic markers for metformin therapy.
The first study on the association of genetic variations of OCT1, OCT2, and MATE1 transporters with gastrointestinal side effects of metformin therapy has been recently published (97). The minor alleles of rs628031 (M408V) and rs36056065 (8 bp insertion), which are two variants of the SLC22A1 gene in strong LD, were significantly associated with the presence of side effects (97). The local increase of drug concentration in the intestinal tissue is proposed as a mechanism of metformin intolerance (98). OCT1 and OCT3 (encoded by SLC22A3 gene) are also expressed in enterocytes (99), and although their role in the intestinal transport of metformin is not yet defined, authors suggest that SNPs in these genes may influence the intestinal metformin uptake and thus induce gastrointestinal side effects (97).
The results of the studies exploring the effect of the only common coding variant in the gene encoding OCT2 (SLC22A2), A270S (rs316019), on metformin clearance, have been contradictory. Two studies found significantly reduced renal clearance of metformin in homozygous variant carriers compared to wild-type homozygotes (100,101). Interestingly, a separate study reported the opposite effect in individuals heterozygous for A270S variant who had higher metformin renal clearance as compared to the reference group (102). Christensen et al. (96) did not find the association between A270S and metformin trough Css or therapeutic response. However, given the importance of OCT2 in metformin pharmacokinetics, further pharmacogenetic studies are needed.
A preliminary study in the incident metformin users from the population-based Rotterdam study cohort assessed the influence of variations in MATE1-encoding gene, SLC47A1, on the HbA1c-lowering effect of metformin (103). From 12 tagging SNPs analyzed, the rs2289669 G>A in intron 10 was associated with greater reduction in HbA1c levels (103). In a latter large-scale prospective clinical study (Diabetes Prevention Program, DPP) an association of rs8065082 SNP in the SLC47A1 gene and lower diabetes incidence in subjects treated with metformin was found (104). This SNP was in high LD with rs2289669 G>A and thus, the DPP study confirmed the findings from a small preliminary study of Becker et al. (103). However, rs2289669 G>A was not associated with metformin renal clearance (93), nor with metformin trough Css (96), and the mechanism underlying the impact of this SLC47A1 SNP remained unclear.
A recent study analyzed the effect of the MATE2-Kgene (SLC47A2) polymorphism on glycemic response to metformin in newly diagnosed T2DM patients (105). A common 5’-UTR variant, g.-130G>A (rs12943590), was associated with enhanced promoter activity and weaker response to metformin, assessed by the relative HbA1c change (105). The study also explored the impact of OCT1, OCT2, and MATE1 variants on the initial metformin response. In line with previous studies (103,104), the intronic rs2289669 G>A SNP in MATE1 gene was associated with higher HbA1c relative change, almost at the level of statistical significance (105).
In addition, other candidate genes which are likely to modulate metformin pharmacokinetics and response are genes encoding OCT3 (SLC22A3) and PMAT (SLC29A4) transporters. Recent in vitro study suggested that OCT3 is implicated in the uptake of metformin in muscle cells and its genetic variants may modulate metformin action (106). Genetic polymorphisms in PMAT showed a tendency of association with lower metformin trough concentration, and could be associated with decreased metformin absorption (96).
Recently, the first genome-wide association study (GWAS) on glycemic response to metformin in T2DM was performed. The study identified a common rs11212617 A>C SNP associated with treatment success in the large GWAS cohort (107), which was replicated in the two additional cohorts (107). The authors proposed that ATM (the ataxia telangiectasia mutated) gene, encoding a serine/threonine protein kinase of the phosphoinositide 3-kinase-related protein kinase (PIKK) family, was the causative gene due to its association with IR, increased T2DM risk, and its role in AMPK activation (107). In general, additional studies are required to identify genes involved in mechanisms of metformin pharmacological action.
Meglitinides
This class of short-acting insulin secretagogues acts by binding to β-cells and inhibiting KATP channel to stimulate insulin release. This is similar to the mechanism of action of the sulphonylureas and both, meglitinides (glinides) and SU, bind at two sites of the SUR1 subunit to inhibit channel activity (108). These two sites are thought to overlap to accommodate the negative charge and the central phenyl ring present in both, SU and meglitinides (109). Repaglinide is specifically designed to stimulate early insulin secretion in the postprandial period after binding to a distinct site on the β-cell (110). Similarly, nateglinide is an amino acid derivative that also induces an early insulin response to meals decreasing postprandial blood glucose levels. Due to their short action, meglitinides have a lower risk to induce hypoglycemia than SU. Furthermore, meglitinides offer an alternative OAD agent of similar potency to metformin, and may be indicated where side effects of metformin are intolerable or where metformin is contraindicated.
Gene polymorphisms associated with the variable meglitinides response are summarized in Table 4. A recent study showed that SLCO1B1gene, which encodes the organic anion-transporting polypeptide 1B1 (OATP1B1), is a major determinant that markedly affects the repaglinide pharmacokinetics (111), consistent with an enhanced hepatic uptake by OATP1B1. SLCO1B1 521T>C SNP appeared to play an important role in the nateglinide pharmacokinetics (112). However, a study done by Kalliokoski et al. (113) found that, in contrast to repaglinide, the nateglinide disposition was not affected by the SLCO1B1 521T>C SNP. Furthermore, the same research group demonstrated that the SLCO1B1*1B/*1B genotype was associated with reduced plasma levels of repaglinide, but had limited effects on the nateglinide disposition (114). A very recent study performed in healthy Chinese population have demonstrated that both, the 521T>C SNP of SLCO1B1 and CYP2C9*3 polymorphism, could significantly affect the nateglinide pharmacokinetics (115). However, these variants were not significantly associated with variations in its glucose-lowering effects and could only partially explain the interindividual variability of nateglinide plasma concentration (115).
Table 4. Summary of the gene polymorphisms involved in the pharmacogenetics of meglitinides.
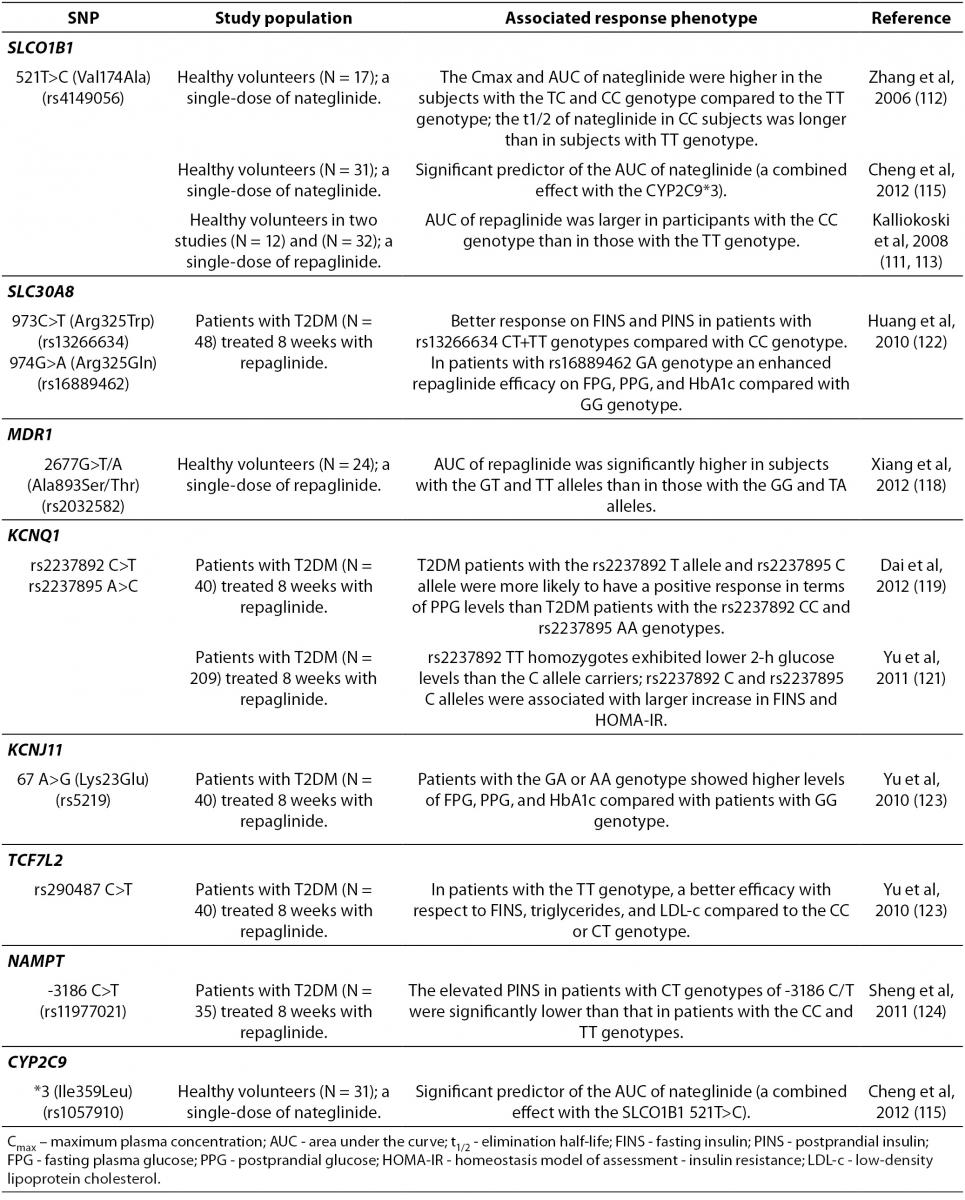
Nateglinide is metabolized by CYP2C9 and moderate dose adjustments based on CYP2C9 genotypes may affect its pharmacokinetics (115) and interindividual variability in its antihyperglycemic effects (70). Repaglinide is metabolized by CYP2C8 and CYP3A4 (116) and, according to clinical studies, CYP2C8*3 carriers have higher clearance than carriers of the wild-type genotypes (70). However, recent study demonstrated that CYP2C8*3 did not appear to affect the repaglinide pharmacokinetics (117).
A recent pharmacogenetic study done in the Chinese population showed that the G2677T/A SNPs of MDR1 gene, encoding P-glycoprotein transporter, were associated with the variability in the repaglinide pharmacokinetics, suggesting that carriers of the MDR1 2677GT and TT allele might be exposed to higher levels of repaglinide (118).
Furthermore, Dai et al. (119) found that rs2237892 (C>T) and rs2237895 (C>A) SNPs, both located in intron 15 of KCNQ1 gene (120),encoding a voltage-gated K+channel expressed in the various tissues including pancreas, were associated with the repaglinide efficacy in Chinese T2DM patients. Diabetic patients carrying the rs2237892 risk C allele had lower fasting insulin levels and HOMA-IR than those carrying the T allele. Furthermore, FPG and HOMA-IR levels were higher in T2DM patients with the rs2237895 risk C allele compared with the carriers of the A allele. Interestingly, following repaglinide treatment, T2DM patients with the rs2237892 T allele and the rs2237895 C allele were more likely to have a positive effect on postprandial glucose levels than patients with the rs2237892 CC and rs2237895 AA genotype (119). In addition, a recent study performed by Yu et al. (121) demonstrated that Chinese T2DM patients, who were rs2237892 TT homozygotes, had lower glucose levels following repaglinide treatment, while the rs2237895 C risk allele in those patients was associated with greater increments in both, fasting insulin and HOMA-IR levels.
Polymorphisms of the zinc transporter solute carrier family 30 member 8 gene (SLC30A8), including rs13266634 (973C>T, Arg325Trp) and rs16889462 (974G>A, Arg325Gln) SNPs, were recently reported to be related to T2DM development and importantly, to the repaglinide efficacy in Chinese T2DM patients (122). Patients with rs13266634 CT+TT genotypes showed decreased fasting and postprandial insulin levels compared to patients with CC genotype, while a significant differences in the decreased fasting and postprandial glucose and HbA1c levels were found between T2DM patients with GA and GG genotype of SLC30A8 rs16888462 SNP. Since SLC30A8 is mainly expressed in the pancreatic β-cells and appears to play a critical role during insulin maturation and release, the authors have speculated that SLC30A8variations influence the zinc disposition and that KATP function, affecting the therapeutic efficacy of repaglinide (122).
Furthermore, the KCNJ11 and TCF7L2 polymorphisms seemed also to be associated with repaglinide efficacy in Chinese T2DM patients (123). Diabetic patients with the GA or AA genotype of the KCNJ11 Lys23Glu SNP showed higher levels of fasting and postprandial glucose and HbA1c following repaglinide treatment than in patients with the GG genotype. On the other hand, in T2DM patients with the TT genotype of TCF7L2 rs290487 (C/T), the drug showed better efficacy with respect to levels of fasting insulin, triglycerides, and low-density lipoprotein cholesterol as compared to carriers of the CC or CT genotype (123).
A recent study performed by Sheng et al. (124) suggested that the -3186C>T SNP of gene encoding nicotinamide phosphoribosyltransferase (NAMPT) was significantly associated with postprandial insulin and total cholesterol levels in Chinese T2DM patients treated with repaglinide. Following the drug treatment, the elevated postprandial insulin levels as well as the total cholesterol levels in patients with CT genotype were significantly lower than in T2DM patients with the CC and TT genotype of the -3186C>T polymorphism (124).
Conclusions
In conclusion, the evidence has been accumulating to show that pharmacogenetics/pharmacogenomics has the potential to improve the management of T2DM and the effective OAD prescribing. Several variants related to drug-metabolizing enzymes, drug-transporters, drug target, and diabetes risk genes have been linked to interindividual differences in the OAD treatment outcomes. As summarized here, significant pharmacogenetic evidence has demonstrated an association between specific gene polymorphisms and interindividual variability in OAD therapeutic and side effects. Identification of drug-genotype interactions in pharmacogenetic studies of the OAD treatment might have clinical implications in the near future resulting in selection of more specific personalized therapy in T2DM. Although benefits from a personalized diabetes care are well established in patients with certain monogenic forms of diabetes, individualized treatment options in the more common polygenic forms of diabetes are also anticipated. However, it is also well understood that the diversity in drug effects cannot be explained by studying the genomic variations only and there are many potential barriers to the translation of pharmacogenetic findings to the antidiabetic treatment. Particularly, epigenomic research that focuses on nongenomic modifications influencing gene expression, may expand the scope of pharmacogenomics towards optimization of drug therapy. Recently introduced “miRNA-pharmacogenomics” that analyzes the polymorphisms in the miRNA regulatory pathway and its association with drug response, would also provide useful information for personalized medicine. Since the pharmacogenetic associations in diabetes that have been reported to date have had limited impact on the individual treatments choice, the value of genetic information in guiding therapeutic decisions in T2DM treatment must be further tested in adequately designed and rigorously conducted clinical trials. With recent scientific and technological advances, pharmacogenomics has a great potential to yield therapeutic advances leading the way towards personalized diabetes care.