Introduction
It is now well established that bone and immune cells are functionally connected. Diverse interactions between bone and immune cells occur within the bone microenvironment. Bone marrow serves as a site for priming naive T cells, recruitment of effector T cells and their proliferation (1,2). Bone and immune cells share the same progenitors residing in bone marrow and are being affected by the same cytokines, influencing hematopoiesis, local immune responses and bone cells as well. In addition, different immune cells such as macrophages, B lymphocytes, mast cells, natural killer cells (NK), etc. have been shown to influence bone cells as well. However, the most powerful players in this regulation are activated T cells. Following successful antigen-specific activation in secondary lymphoid tissues, T cells begin to produce numerous pro-inflammatory cytokines such as interleukin (IL)-2, -4, -5, -6 and -8, tumor necrosis factor-α (TNF-α), transforming growth factor-β (TGF-β), granulocyte-macrophage colony stimulating factor (GM-CSF), interferon γ (IFN-γ), etc. (3). These pro-inflammatory cytokines are soluble mediators with pleiotropic effects on different types of cells. Interestingly, they affect bone cells within a particular bone microenvironment. The pro-inflammatory cytokines can be classified either based on the primary source of their production, i.e. the type of immune cell or their specific targets in bone, i.e. bone cells and thus the process they affect, i.e. bone resorption or formation. Although these cytokines can influence both, the bone forming cells (osteoblasts) and the bone resorbing cells (osteoclasts), the majority of existing data is focused on the influence of immune cells on osteoclasts and thus on bone resorption. Nevertheless we have to take in account that pro-inflammatory cytokines such as TNF-α and IL-17, released from activated T cells, can target osteoblasts as well. The result of such influence is the increased production of pro-inflammatory cytokines and RANKL in osteoblasts and thus the contribution of these cells to bone resorption (4). Depending on their influence on osteoclast differentiation, activation or survival, the pro-inflammatory cytokines can roughly be divided into osteoclastogenic, if they stimulate osteoclasts, or anti-osteoclastogenic, if they inhibit these unique bone-resorbing cells. Initially, the interplay between immune cells and osteoclasts has been observed in clinical settings of increased bone resorption following glucocorticoid therapy and in typical inflammatory bone diseases, such as rheumatoid arthritis (RA) and periodontal disease. The cross-talk between immune and bone cells via cytokines, chemokines, growth factors, signaling molecules and transcription factors, has begun to be intensively studied with the onset of osteoimmunology. This terminus was first mentioned in 2000, when Arron pointed out the importance of studying both, the skeletal and immune system, as an integrated entity (5). The milestone for osteoimmunology however, dates far before that, already in 1972, when Horton et al. found a new soluble factor in the supernatant from cultures of stimulated human peripheral blood leukocytes that was capable to trigger in vitro bone resorption (6). More than 20 years later, four different groups of researchers independently characterized the receptor activator of nuclear factor κB ligand (RANKL), an essential molecule that controls osoteoclastogenesis and thus bone resorption. The first two groups have found strong upregulation of RANKL in immune cells such as antigen-stimulated T cells and in mature dendritic cells (7,8) and the second two groups in bone marrow stromal cells (9,10). These findings pinpointed the important role of RANKL within both, skeletal and immune system, leading to first speculations that both systems might be functionally connected. However the main stimulus giving rise to osteoimmunology was the study of Takayanagi et al. in 2000, where they showed in vitro bone protecting action of IFN-γ (11). Every time our T cells are activated, they release RANKL, the osteoclast-stimulating molecule inducing bone resorption. Interestingly, IFN-γbinds to its receptor on osteoclasts, degrades RANKL signaling and thus inhibits the activation of osteoclasts and protects our bones from being resorbed. This cytokine is produced predominantly by NK and natural killer T (NKT) cells involved in the innate immune response, and by CD4+ Th1 and CD8+ cytotoxic T lymphocyte (CTL) effector T cells, once antigen-specific immunity develops (12).
As already mentioned the role of pro-inflammatory cytokines has been most extensively studied in RA (13). With the advancement of osteoimmunology the involvement of these soluble factors in the pathogenesis of bone diseases that have traditionally not been considered as inflammatory, such as osteoporosis (OP) and osteoarthritis (OA), has also been suggested (14,15).
The scope of this review is to summarize the current evidence on the influence of pro-inflammatory cytokines on osteoclasts and thus on the process of bone resorption. In the first part, the bone microenvironment, involving osteoclasts, osteoblasts and their connection to immune cells, will be addressed, while in the second part the osteoclastogenic, anti-osteoclastogenic and mixed (dual) effects of pro-inflammatory cytokines will be reviewed. Finally, up to date evidence on the role of pro-inflammatory cytokines in OP and OA will be summarized, with the aim to show that researching such complex bone disorders is almost impossible without considering skeletal and immune system in an integrated way.
Bone microenvironment
The interplay between bone and immune cells
Bone and immune cells are very similar meaning they have common progenitors in bone marrow, they require common receptors for cell signaling, and immune cells have shown the ability to differentiate into osteoclast as well.
Osteoclasts, macrophages and dendritic cells all derive from the same progenitors, i.e monocyte-macrophage lineage (16). Besides RANKL and macrophage colony stimulating factor (M-CSF), which are essential for commitment of the common precursor to the osteoclast lineage and survival of generated osteoclasts, numerous cytokines are also able to influence osteoclast differentiation and/or function (17). RANKL, when signaling via its receptor activator of nuclear factor κB (RANK) on osteoclasts, requires expression and activation of costimulatory molecules, containing immunoreceptor tyrosine-based activation motif (ITAM) domains. ITAMs are important components of numerous immune receptors. They are also present in DNAX adaptor protein 12 (DAP12) and Fc receptor γ (FcRγ) adapter molecules. The receptors that associate with DAP12 are the triggering receptor expressed by myeloid cells-2 (TREM-2) and the signal regulatory protein β1 (SIRP β1) (18). Receptors that associate with FcRγ adapters in myeloid cells are the osteoclast associated receptor (OSCAR) and the paired Ig-like receptor A (PIR-A). Mice lacking DAP12 and FcRγ are severely osteopetrotic (18,19). Interestingly, OSCAR can partially rescue the abnormal cytoskeleton of the osteoclasts in DAP12-/- mice (20) and was recently shown to bind to fibrillar collagen and promote osteoclastogenesis (21). These data suggest OSCAR as important immunoreceptor in osteoclastogenesis.
Furthermore, for osteoclast precursors to turn into mature resorbing osteoclasts, the interaction between members of the immunoglobulin superfamily, namely the type-1 membrane glycoprotein CD200 and its receptor (CD200R), are needed. Osteoclast numbers decreased and the bone mass was increased in CD200 deficient mice (22). During different phases of their development, osteoclasts express specific molecules such as RANK, calcitonin receptor, β3 integrin, OSCAR, cathepsin K and tartrate-resistant acid phosphatase (TRAP), which are involved in osteoclast differentiation, function and survival (23).
Immune cells have shown the ability to differentiate into osteoclasts as well. Dendritic cells, which are professional antigen presenting cells able to efficiently activate naïve T lymphocytes, can differentiate into osteoclasts in vitro during their early stage of development (24). This process is stimulated by pro-inflammatory cytokines IL-1β and TNF-α and is inhibited in the presence of IFN-α (25). Moreover, osteoclasts from myeloma patients contain nuclei with translocated chromosomes of myeloma B-cell clone origin, suggesting that B-lymphoid lineage cells might also give rise to osteoclasts (26).
Osteoblasts, the bone forming cells, originate from bone marrow-residing multipotent mesenchymal stem cells. Osteoblasts are one of the major sources of RANKL and in this manner they control bone resorption. Interestingly, they influence immune cells as well. Osteoblasts are critical regulators of the hematopoietic stem cells (HSC), located in a specialized structure within bone marrow, the niche, from where immune and other blood cells derive (27). This regulation occurs via annexin II, an osteoblast derived protein. It has been shown that bone marrow of annexin II-deficient mice contains decreased number of hematopoetic stem cells (28). HSC can remain dormant or replicate to either self-renew or differentiate into multipotent progenitor cells capable of differentiating into lymphoid or myeloid precursors. In addition, the production of IL-10 by osteoblasts has also been shown to promote the self-renewal of HSC in their bone marrow located niches (29). Primary osteoblast lineage cells have been shown to produce M-CSF, granulocyte colony stimulating factor (G-CSF), GM-CSF, IL-1, IL-6, lymphotoxin, TGF-β, TNF-α, leukemia inhibitory factor (LIF), and stem cell factor (SCF), which play a role in hematopoiesis and many of them are involved in osteoclast development as well (30).
RANK/RANKL/OPG system
Discovery of the molecular triad RANK/RANKL/osteoprotegerin (OPG) and characterization of their central role in the interplay between immune and bone cells, have contributed significantly to the emergence of the osteoimmunology (31). RANKL, a member of the TNF superfamily, is a potent stimulator of both, osteoclast formation and their bone-resorbing activity (31,32). Upon binding to its receptor, RANK located on osteoclasts, RANKL signaling increases differentiation and activation of the osteoclasts resulting in expression of osteoclast specific molecules. Although it has been proposed for a long time that the main source of RANKL are stromal and osteoblastic cells, a very recent study by Nakashima et al. proved that the main RANKL production site resides within osteocytes (33). OPG is a secreted TNF receptor superfamily member acting as a decoy receptor molecule for RANKL, thereby counteracting its osteoclastogenic activity. It is produced by a variety of cells, including stromal cells, B lymphocytes and dendritic cells (34). RANK or RANKL knockout mice have significant osteopetrosis with no osteoclasts. In contrast, animals lacking OPG develop severe OP due to a response to enhanced osteoclastogenesis (35). Mutations in humans causing overexpression of RANK are associated with familial expansile osteolysis and expansile skeletal hyperphosphatasia (36,37). Anti-RANKL specific antibody, denosumab has been recently approved in clinical practice for the treatment of OP (38).
Numerous osteoclastogenic factors including hormones, cytokines and growth factors, that are produced physiologically or excessively during inflammation, are now believed to exert their primary activity on osteoclasts by modulating the RANK/RANKL/OPG axis (39). Upon activation T cells produce both, RANKL and pro-inflammatory cytokines and can thus influence osteoclastogenesis (40-42).
The influence of pro-inflammatory cytokines on osteoclasts
Several systemic and local factors are known to influence osteoclasts and their bone resorption ability. In a physiologic state, the osteoclast activity is highly balanced by those factors. In pathological conditions, such as excessive activation of the immune system, their activity becomes deregulated due to additional pro-inflammatory cytokines, produced mainly by activated T cells (43). As already mentioned, the pro-inflammatory cytokines are osteoclastogenic if they have shown stimulation of osteoclast differentiation and/or activity, or anti-osteoclastogenic, if their ability to inhibit osteoclasts has been proved (44). Based on the current evidence, pro-inflammatory cytokines such as IL-1, IL-6, IL-8, IL-11, IL-17, TNF-α etc. are considered osteoclastogenic, and IL-4, IL-10, IL-13, IL-18, IFN-γ, IFN-β etc. anti-osteoclastogenic. Th17, a special subtype of T helper cells, producing IL-17 and RANKL, are classified as osteoclastogenic, while the classical Th1 and Th2 cells are considered anti-osteoclastogenic, due to their production of IFN-γ (Th1) and IL-4 (Th2) (45,46). Some of the pro-inflammatory cytokines, such as IL-7, IL-12, IL-23, and IL-6 and TGF-β as well, have been shown to possess dual, osteoclastogenic and anti-osteoclastogenic properties. It seems that their net effect depends on the specific pathophysiological condition of bone in which they are being studied in vivo, while in experiments in vitro it depends on developmental stage of osteoclasts (43,47,48). The determination of their exact role in bone microenvironment is even more difficult, as synergistic and antagonizing effects on osteoclasts of these cytokines have been observed (49-56). As already mentioned, the pro-inflammatory cytokines can target osteoclasts directly or indirectly by modulating the RANK/RANKL/OPG system. The osteoclastogenic effect of these cytokines can be assessed by determining the expression of osteoclast-specific genes encoding RANK, calcitonin receptor, β3 integrin, OSCAR, cathepsin K and TRAP, being involved in osteoclast differentiation, bone-resorbing activity and survival (23). The schematic representation of the influence of the pro-inflammatory cytokines on osteoclast is shown in Figure 1.
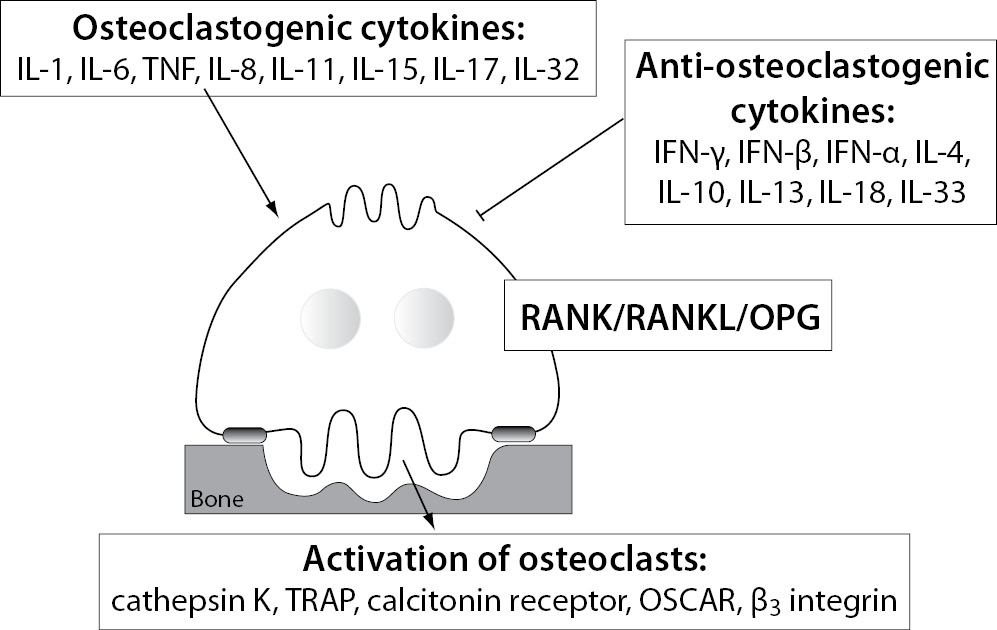
Figure 1. Different influences of pro-inflammatory cytokines on osteoclasts.
IL - interleukin; TNF- tumor necrosis factor; IFN - interferon; RANK - receptor activator of nuclear factor κB; RANKL - RANK ligand; OPG - osteoprotegerin; TRAP - tartrate-resistant acid phosphatase; OSCAR - osteoclast associated immunoreceptor.
Cytokines with stimulating effects on osteoclasts (osteoclastogenic cytokines)
Mechanisms of action of osteoclastogenic pro-inflammatory cytokines, the evidence on their opposite effects, and synergy or antagonism with other cytokines are summarized in Table 1.
Table 1. Osteoclastogenic cytokines.
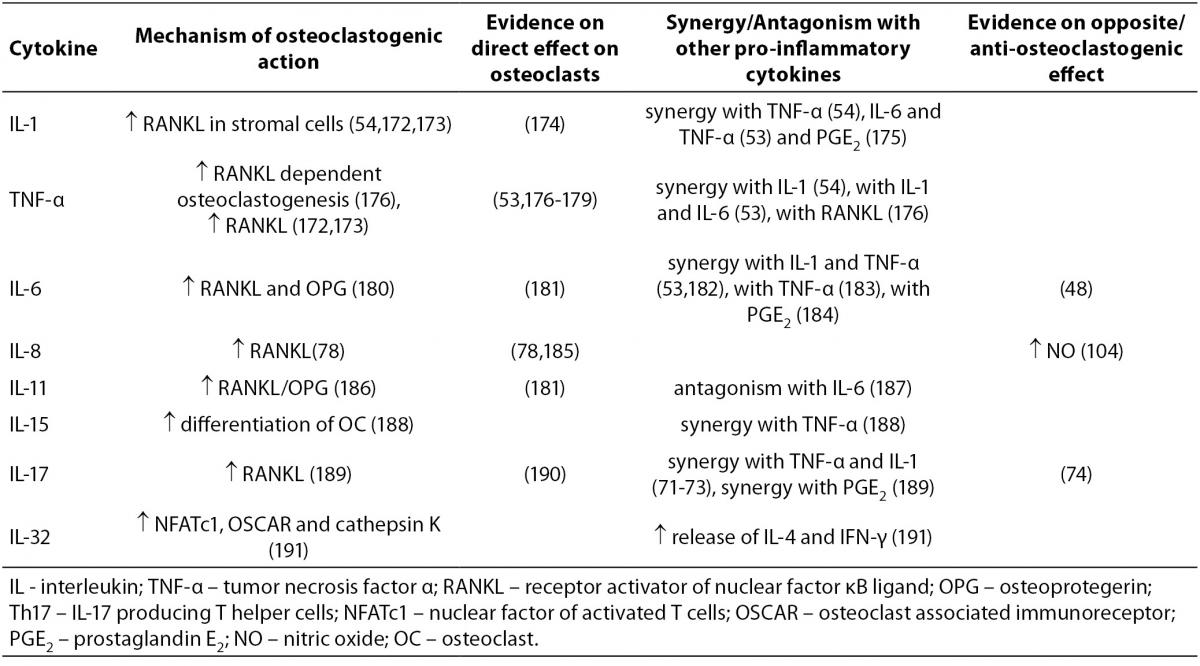
IL-1, existing in a form of IL-1α and IL-1β polypeptides, is produced by macrophages and is a potent stimulator of bone resorption, both in vitro and in vivo (57-61). Similarly to IL-1, TNF is also represented as a family of two related polypeptides α and β that are both potent stimulators of osteoclastogenesis (60,62,63). TNF-α is produced by a variety of immune cells, primarily by macrophages, but also by NK cells, mast cells and T (Th1) and B lymphocytes. Anti-TNF-α antibody is used for the treatment of RA (12). IL-6, produced by T cells (Th2), dendritic cells and macrophages, is a pivotal cytokine in acute and chronic inflammation. Interestingly, stromal cells and osteoblasts can also produce nanomolar quantities of IL-6 in response to TGF-β, IL-1 and TNF-α (64). The blocking of IL-6 signaling is effective in controlling RA symptoms (65), as well as in vivo and in vitro blocking of osteoclast formation (66). Interestingly, data on direct inhibition of osteoclast differentiation by IL-6 are also available (48). IL-11 is produced in bone marrow stromal cells and is required for normal bone remodeling (67). In vitro studies have shown that it stimulates osteoclast formation and bone resorption(68). Mice being deficient in specific IL-11 receptor have increased trabecular bone mass due to decreased bone resorption (69). IL-17 exists as a family of six distinctive cytokines (IL-17A - F) that are produced by a distinct lineage of T helper lymphocytes, the Th17 cells and are able to exert high osteoclastogenic activity (70). In a synergy with TNF-α and IL-1, IL-17 has been causatively implied in the bone destruction observed in RA (71,72). IL-17A inhibition in an antigen-induced arthritis model, prevented joint and bone destruction and decreased the levels of RANKL, IL-1β, and TNF-α within pathologic lesions (73). Recent study however showed, that the osteoclastogenic effect of IL-17A depends on its concentration, as only high concentrations of IL-17A were able to suppress osteoclastogenesis in vitro (74). Like IL-17, IL-15 can also be produced by T lymphocytes, although its main sources are monocytes/macrophages and dendritic cells (75) and is involved in bone destruction in RA as well (76). Mice deficient in IL-15Rα, the specific IL-15 receptor subunit have increased bone mineral density (BMD) and decreased numbers of osteoclasts (77). IL-8 belongs to a group of chemokines and is, unlike other osteoclastogenic cytokines that we have described up to this point, produced not only by T cells and macrophages but also by osteoclasts themselves. It has been shown to play an osteoclastogenic role in metastatic bone disease (78). IL-32 is a relatively newly described cytokine showing typical properties of a pro-inflammatory mediator, as it stimulates TNF-α, IL-1β and IL-8 production. It exerts complex osteoclastogenic effects, such as promoting osteoclast differentiation but not maturation of osteoclasts (79).
Cytokines with inhibiting effects on osteoclasts (anti-osteoclastogenic cytokines)
The mechanisms of action of anti-osteoclastogenic pro-inflammatory cytokines, the evidence on their functional effects, as well as synergy or antagonism with other cytokines, are summarized in Table 2.
Table 2. Anti-osteoclastogenic cytokines.
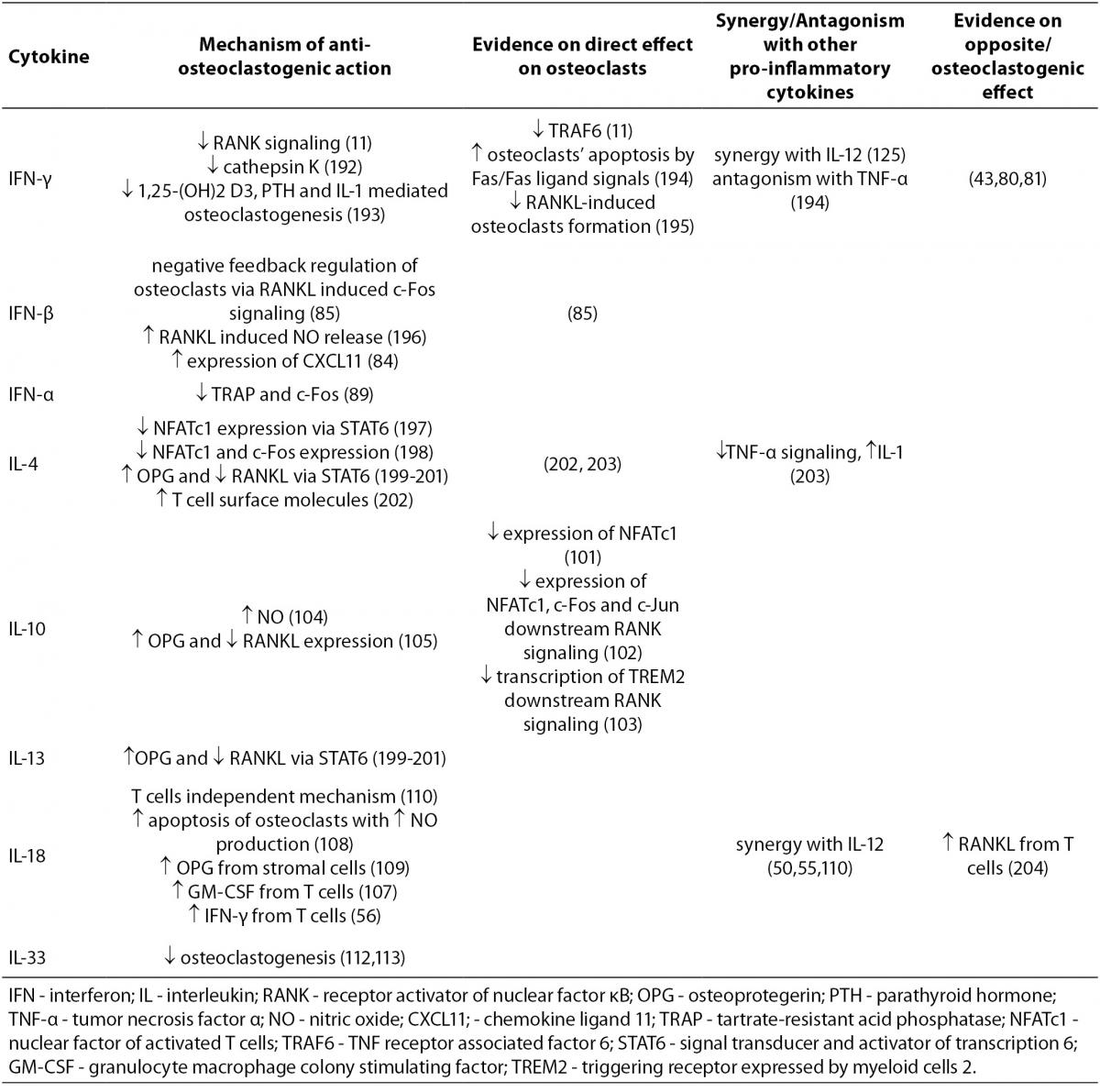
IFN-γ, a type II interferon, which is produced by Th1 lymphocytes and NK cells, is a potent inducer of antigen presentation and therefore of T cell activation. As previously mentioned, the discovery of IFN-γ anti-osteoclastogenic effect in vitro has initiated development of osteoimmunology (11). In vivo effects of IFN-γ on bone tissue are more difficult to categorize and often indicate its opposite, osteoclastogenic effect as well (43,80,81). It seems that IFN-γ functions as an anti-osteoclastogenic cytokine in physiological conditions of bone turnover as proven by lower BMD in nude and IFN-γ receptor deficient mice (43,81). However, in pathological bone turnover such as in postmenopausal OP, inflammation or bacterial infection, the net effect of IFN-γ is biased towards bone resorption via antigen driven T cell activation and RANKL production (43). In rats, intraperitoneal IFN-γ injections induced osteopenia (82), while in humans this cytokine has been reported to be efficacious in the treatment of osteopetrosis as it restores bone resorption (83).
Comparing to IFN-γ, there are fewer studies published on the effects of type I interferons, IFN-α and IFN-β. However, the existing data on the anti-osteoclastogenic activity of IFN-β are quite uniform (84). Namely, mice that are deficient in IFN-β exhibit severe osteopenia accompanied by enhanced osteoclastogenesis (85). In order to suppress the anti-osteoclastogenic effects of IFN-β, osteoclast precursors have been shown to upregulate the expression of the suppressors of cytokines signaling (SOCS) (86). Recently, the upregulation of IFN-β expression has been suggested as a basic molecular mechanism needed for the efficacy of vitamin D (1α,25(OH)2D3) used to treat human bone diseases (87). In animal models of RA, the application of IFN-β resulted in significantly reduced cartilage and bone destruction accompanied by downregulated expression of TNF-α and IL-6, as well as increased expression of IL-10 (88).
There are even much more limited data on IFN-α, showing its anti-osteoclastogenic effects in vitro (89). In vivo no effect of IFN-α on bone turnover could be detected in rats (90), however in mice models of systemic lupus erythematosus it was shown to decrease bone resorption (91). Children receiving IFN-α therapy due to chronic hepatitis B have been shown to have higher femoral BMD (92).
IL-4 and IL-13 are anti-osteoclastogenic cytokines, moreover they have shown influence on osteoblast as well (93,94). Transgenic mice overexpressing IL-4 develop an osteoporotic phenotype (95) that may result from both, the inhibition of osteoclast formation and their activity (96,97), as well as the inhibition of osteoblasts and bone formation (94). Both interleukins have also been shown to inhibit IL-1 stimulated bone resorption by decreasing the activity of cyclooxygenase-2 (COX-2) and thereby the production of prostaglandins (98). Additionally, IL-4 has the ability to inhibit osteoclastogenesis being physiologically stimulated by RANKL or pathologically by TNF-α (99).
IL-10 is produced by monocytes, T cells and B cells (100) and inhibits not only osteoclastogenesis (101-105) but also osteoblastogenesis and the onset of mineralization (106).
Besides IL-17 and IL-23, IL-18 has been also found to be increased in inflammation sites in RA (56). Although it belongs to the IL-1 superfamily, and is also structurally similar to IL-1, it has been shown to exert its anti-osteoclastogenic properties via various mechanisms (56,107-110). It is produced by macrophages and other cells, also osteoblastic ones. IL-18 overexpressing transgenic mice have decreased numbers of osteoclasts and decreased bone mass (111). IL-33 is another member of the IL-1 superfamily, and recent data suggest that similarly to IL-18, it has anti-osteoclastogenic effects (112,113).
Cytokines with dual (opposite) effects on osteoclasts
The mechanisms of action of pro-inflammatory cytokines with mixed, osteoclastogenic and anti-osteoclastogenic effects are summarized in Table 3.
Table 3. Cytokines with osteoclastogenic and anti-osteoclastogenic properties.
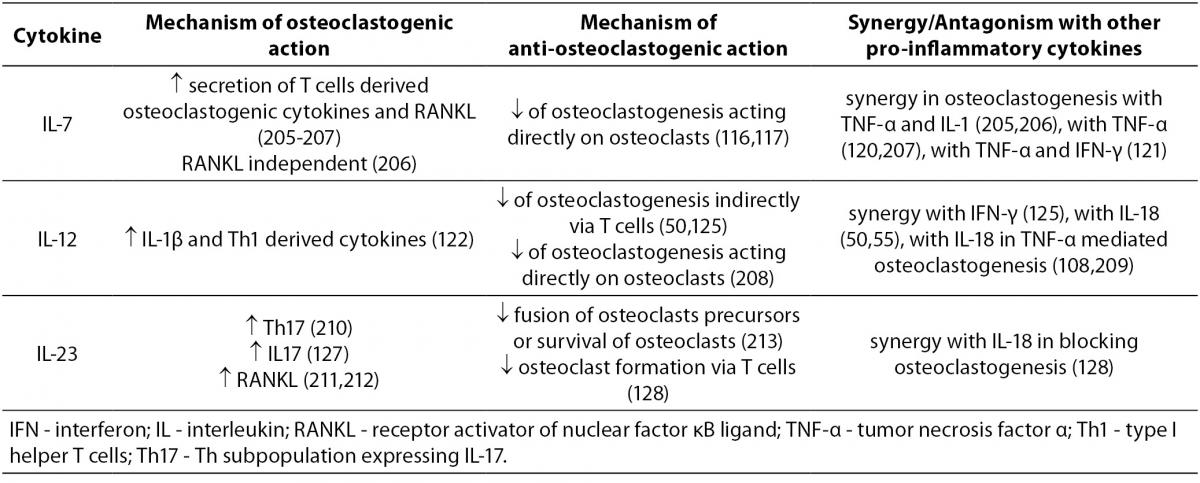
IL-7 is produced by stromal cells in red bone marrow and thymus, keratinocytes, follicular dendritic cells and other non-hematopoietic cells. The production of IL-7 was enhanced in ovariectomized (OVX) mice where it stimulated osteoclastogenesis (114). Another study found that in IL-7 deficient mice, as compared to wild-type animals, the numbers of osteoclasts increased and the trabecular bone volume decreased; however both types of mice displayed similar bone loss following OVX (115). Addition of IL-7 to murine bone marrow cells cultured in the presence of M-CSF and RANKL, inhibited osteoclast formation as well (116). Recently, mice overexpressing human IL-7 in their osteoblast lineage were found to have increased trabecular bone volume in vivo and decreased osteoclast formation in vitro (117). In thymectomized women the levels of IL-7 and RANKL were increased compared to controls, however their RANKL/OPG ratios and indices of bone metabolism were not affected (118). In humans, IL-7 has shown its osteoclastogenic nature in psoriatic arthritis (119) and in solid tumors bearing patients (120), while in those suffering from RA it was suggested to contribute to the perpetuation of Th1 and TNF-α mediated pro-inflammatory immune responses (121).
IL-12 is another controversial cytokine in terms of its anti-osteoclastogenic/ osteoclastogenic effects (122). It is produced by myeloid cells, i.e. dendritic cells and macrophages and induces differentiation and activation of Th1 lymphocytes and their subsequent production of IFN-γ and GM-CSF (123,124). There are conflicting results on the involvement of IFN-γ in IL-12 mediated osteoclastogenesis inhibition as well (50,125).
IL-23 is produced by dendritic cells and macrophages. It plays a role in bone destruction in RA, as shown in mice models of RA, with animals being deficient for either IL-17 or IL-23 (70,126), and in rat models of collagen-induced arthritis, where animals were treated with anti-IL-23 antibodies (127). However, in non-pathological conditions IL-23 functions in an opposite, anti-osteoclastogenic way (128).
Transforming growth factor β
TGF-β, an ubiquitous growth factor, is not a pro- but an anti-inflammatory cytokine, playing an important and complex role in bone remodelling by influencing bone resorption, formation and the production of certain pro-inflammatory cytokines (47,49). It is produced by many different cells, including macrophages and exists in at least three isoforms, TGF-β1, TGF-β2 and TGF-β3. Recently, the role of TGF-ß1 in coupling bone formation and resorption has been suggested (129). Its effects on osteoclasts cultured in vitro greatly depend on numerous factors, such as: differentiation stage of the starting cell population, cell density, the presence or absence of serum in the cell culture medium, concentration of added TGF-ß1, and whether its effect was studied on isolated cells or in co-cultures. In vivo the presence of other cytokines such as IL-1, IL-6, TNF-α, IFN-γ, prostaglandin E2 (PGE2) etc. and environment as such determine the exact outcome of TGF-ß1 activity (47,51).
The pro-inflammatory cytokines and bone diseases
In many pathological conditions, activated immune cells such as T lymphocytes, macrophages and dendritic cells produce a variety of pro-inflammatory cytokines which in turn modulate osteoclast activity and bone resorption as already described above. In continuation we will provide actual data on the roles of certain pro-inflammatory cytokines in two common bone diseases, OP and OA.
The role of pro-inflammatory cytokines in osteoporosis
Osteoporosis has been traditionally considered an endocrine disease resulting mainly from the estrogens decline after menopause. This change affects bone remodeling, leading to impaired microarchitecture of bone tissue and consequently to higher risk of fractures. Since solely the endocrine point of view does not fully explain the pathogenesis of OP, the osteoimmunological approach suggests that activated T lymphocytes can contribute to menopausal changes in bone remodeling by producing pro-inflammatory cytokines such as TNF-α, IL-1, IL-6, IL-7 and IFN-γ (130,131). This statement has caused a paradigm shift in the pathogenesis of OP, which can now be also classified as an inflammatory disease (4, 15,132). The effects of pro-inflammatory cytokines have been initially studied in OVX mice which represent an experimental model of postmenopausal OP. Nude mice that are deficient in T cells were found to be resistant to bone loss following acute estrogen decline, artificially caused by OVX. It has been proposed that this was due to the lack of CD4+ subpopulation of T lymphocytes (130). Another mechanism of bone loss due to estrogen decline is the enhanced TNF-α production by activated T cells (133,134). Mice deficient in IL-1 receptor (135), TNF-α (133,136) or IL-6 (68) did not lose bone mass following OVX, while no difference in bone loss could be found between IL-7 deficient mice and the wild type animals (115). However, there are also studies showing the opposite effects. Namely, no influence on trabecular bone loss has been observed in mice lacking functional T lymphocytes after OVX (137), and additionally nude mice can develop OP as well (138). Studies in humans have shown higher T cell activity and increased levels of IL-1, TNF-α and IL-6 in postmenopausal osteoporotic women (130,139-143). Higher expression of RANKL/OPG and M-CSF in patients with fragility fracture has also been reported (144). Furthermore, increased serum levels of IL-6 were proved to account for up to 34% of the total variance of BMD change within the first few years after the onset of menopause (145). In Figure 2, the connections of the pro-inflammatory cytokines with OP phenotype in patients with low-energy femoral neck fractures are presented according to results of our recently published study (146). In this study we found higher expression of genes encoding for IL-1α, IL-6, RANKL and RANK in osteoporotic bone tissue. Additionally, we have found two genetic variants in IL-1α gene showing possible influence on osteoporotic phenotype, such as osteoporotic fracture (147). It has been suggested that activated T cells could contribute to enhanced osteoclastogenesis in two ways, first by increasing the production of bone resorbing cytokines, especially TNF-α and RANKL, and secondly, by increasing the numbers of osteoclast precursors (139,148). Bone cells isolated from untreated postmenopausal women showed higher mRNA expression for IL-1, TNF and IL-6 in comparison to those taken from women on estrogen replacement therapy (149). A blockade of both, TNF and IL-1 reduced bone resorption in postmenopausal osteoporotic women (150). Interestingly, Canadian multicentre osteoporosis study showed that treatment with COX-2 inhibitors was associated with higher BMD in postmenopausal women not taking estrogen (151). When looking at the extent of IFN-γ production, no differences between postmenopausal osteoporotic and healthy women were found (152). As already mentioned in case of IFN-γ, differences exist in effects of pro-inflammatory cytokines on osteoclasts depending on whether the bone is being remodelled under physiological or pathological conditions. In pathological conditions such as inflammation, infection and estrogen deficiency, Gao et al. have shown that the net balance between osteoclastogenic and anti-osteoclastogenic effect of IFN-γ is shifted towards osteoclastogenesis and bone resorption (43). This is due to predominant indirect effect of IFN-γ that presents enhanced antigen presentation by IFN-γ to T cells leading to T cells’ activation, proliferation and production of osteoclastogenic cytokines, such as RANKL and TNF-α. In vivo, this indirect (via RANKL) osteoclastogenic activity overcomes the direct suppressive action of IFN-γ on osteoclast precursors, leading to a net bone loss. Similarly, IL-6 does not seem to play an osteoclastogenic role under physiological conditions such as the estrogen-replete state (131).
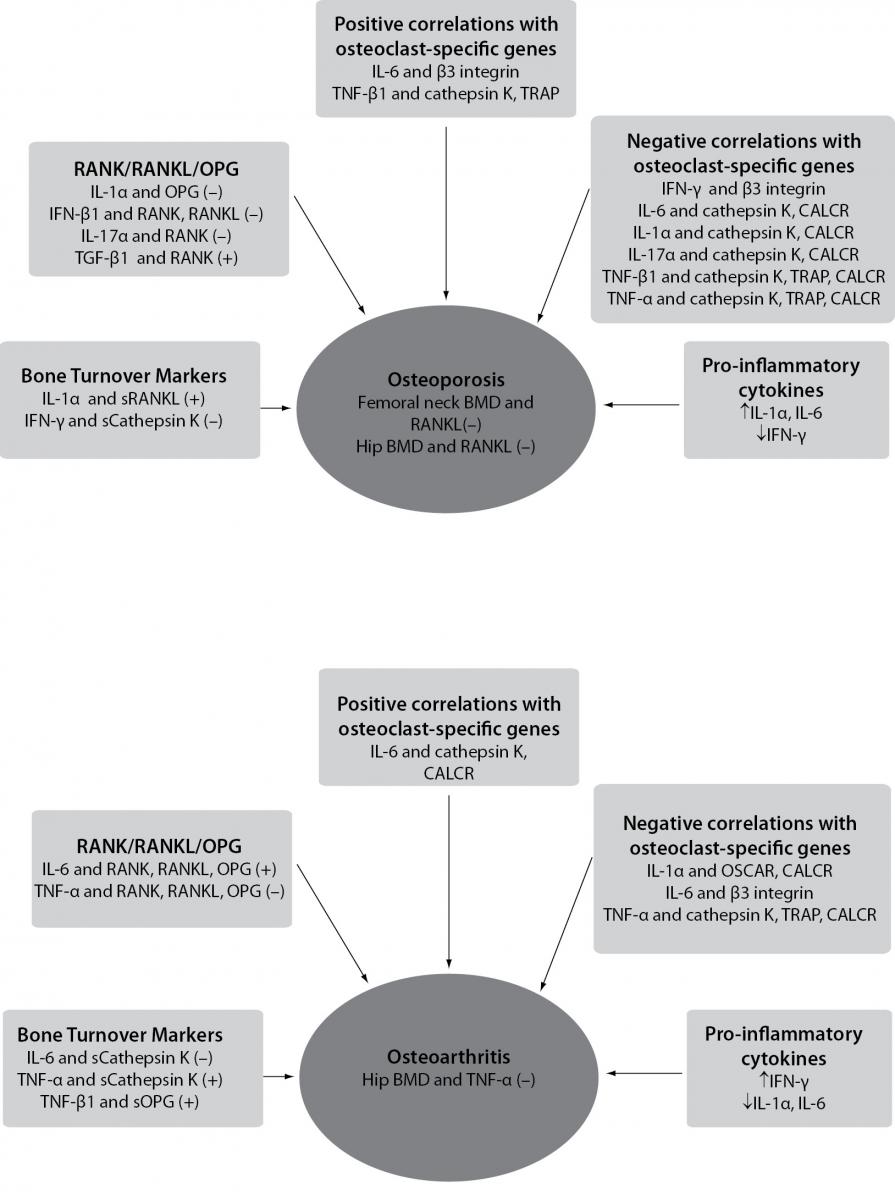
Figure 2. Schematic representation of correlations between pro-inflammatory cytokines, osteoclasts-specific genes, bone mineral density and levels of bone turnover markers in osteoporosis and osteoarthritis, according to results of Zupan et al. (146).
(–) - denotes negative correlation; (+) - denotes positive correlation; BMD - bone mineral density; RANK/RANKL/OPG - the most significant correlations of pro-inflammatory cytokines with RANK, RANKL and OPG genes are shown; sCathepsin K - cathepsin K serum levels; sOPG - OPG serum levels; sRANKL - RANKL serum levels; OSCAR - osteoclast-associated immunoglobulin-like receptor; CALCR - calcitonin receptor; TRAP - tartrate-resistant acid phosphatase.
Rodent models were used to show that TGF-β1 and TGF-β2 prevent bone loss following OVX (153) and that estrogen upregulates the expression of TGF-β (154). There is also evidence on the involvement of TGF-β into a mechanism by which estrogen deficiency upregulates the production of IFN-γ (155,156). In humans, long-term in vivo estrogen treatment has been shown to increase serum levels of TGF-β1 and TGF-β2 (157).
The role of pro-inflammatory cytokines in osteoarthritis
The role of pro-inflammatory cytokines such as IL-17, IL-1 and TNF-α in bone loss has been most intensively elucidated in RA and periodontal disease. In contrast to RA, OA has long been considered an age-related degenerative disease of cartilage with accompanying pathological changes in subchondral bone. Since the concept of synovial inflammation contributing to OA was suggested, numerous studies have shown that the inflamed synovium and activated synovial macrophages importantly promote the osteoarthritic pathology (14,158,159). These results provided the missing inflammatory component to the pathology of this disease. Unlike in OP, where the influence of pro-inflammatory cytokines has been focused on bone cells, the most intensively studied targets of these soluble factors in OA are synovial cells and chondrocytes. Synovial biopsy specimens taken from patients with early OA or RA are similar in cell morphology and cytokine spectra, although the percentage of macrophages, T and B lymphocytes, as well as the levels of TNF-α and IL-1, are generally lower in OA (160). TNF-α and IL-1 have been suggested as key players in pathogenesis of OA. Namely, they have been shown to promote their own production, induce synovial cells and chondrocytes to produce other pro-inflammatory cytokines such as IL-6 and IL-8, as well as to stimulate protease and prostaglandin production (158,161,162). Animal models and human studies of OA have shown decreased cartilage damage following anti-IL-1 antibody therapy or gene transfer of IL-1Ra, the IL-1 receptor antagonist (163,164). In mice with OA, being deficient in IL-1 the pathology of OA was reduced (162,165). However, another study in IL-1 deficient mice found the opposite results (166), indicating a more complex role of IL-1 in OA. In our opinion this disrepancy could be due to different models of OA in animals, whether IL-1 or just IL-1β was studied or similarly to IFN-γ, IL-1 could play dual role depending on the presence of other cytokines. Interestingly, IL-4, IL-10 and IL-13 that are classified as anti-osteoclastogenic cytokines according to their influence on bone, have fully proven their anti-inflammatory role in OA. They were increased in synovial fluid of OA patients and were shown to inhibit a number of pro-inflammatory cytokines, such as IL-1β, TNF-α, PGE2, etc. (161).
Synovial macrophages play an important role in perpetuating the production of pro-inflammatory cytokines and mediating osteophyte formation and fibrosis in subchondral osteoarthritic bone (167). The latter two processes may also be under control of TGF-β (168). Namely, the inhibition of TGF-β markedly reduced both, fibrosis and osteophyte formation. The impaired TGF-β signaling then leads to chondrocyte hypertrophy and results in osteoarthritic pathology of cartilage as well (168,169).
Data on the influence of pro-inflammatory cytokines on osteoarthritic bone are scarce. A recent study pinpointed the increased gene expression of TGF-β (144). Our own data have shown higher expression of TGF-β1 receptor and the anti-osteoclastogenic cytokine IFN-γ in bone tissue samples of osteoarthritic patients, and connections of the pro-inflammatory cytokines with BMD, serum levels of bone turnover markers and osteoclast-specific genes as shown in Figure 2 (146). Another study in primary hip osteoarthritic patients showed positive correlations between IL-8 and bone resorption marker CTX, C terminal telopeptide of collagen type I in both, serum and synovium (170). However most of the published data indicate that in human OA, RANKL, released from activated T cells and synoviocytes, is the principal mediator of bone destruction. Additional proof for that is the fact that RANKL inhibition does not diminish inflammation but effectively prevents bone loss at the site of inflammation (35). Also, in randomized controlled clinical trials no effect in preventing bone loss in patients with RA following IFN-γ treatment could be found (171).
Conclusion
Thanks to recent advances in osteoimmunology, remarkable progress has been made in elucidating dynamic cross-talk between bone and immune cells that takes place in a complex bone microenvironment. Osteoimmunology has postulated a new approach to studying bone diseases such as OP and OA that were traditionally not considered to be inflammatory. Data reviewed in this paper provide an insight into the influence of pro-inflammatory cytokines on osteoclasts and thus on the bone resorption. They clearly show that T cell-derived cytokines can exert different effects on osteoclasts very much depending on whether a study was performed in vitro or in vivo. A huge step forward to elucidation of interactions between immune and skeletal system has been made by using animal models such as OVX mice and rodent models of OA. However, further studies in humans will be critical for clearly understanding the roles of pro-inflammatory cytokines in bone loss with the aim of raising novel therapeutic strategies for these kinds of patients.