Introduction
Continuous technological innovation requires the laboratory to deal with frequent changes in methodology and instruments. Some of these changes are motivated by manufacturer´s improvements. Moreover, sometimes they are promoted by set requirements, which depend on the clinical needs of the diagnostic test.
According to Clauses 5.5.1 to 5.5.3 of ISO 15189 (1), accredited laboratories may modify methods. Such modifications require the accredited laboratory to have a flexible scope of accreditation. The main consequence of a flexible scope and the benefit to the laboratory is the flexibility to modify methods, validate the changes and apply them without having to ask the Accreditation Body for extensions to the scope as in the called fixed scopes of accreditation are required (2-4).
Historically, accreditation has been based on so called fixed scopes. This system for defining the scope obliges that an evaluation of laboratory competence should be carried out each time when an additional activity is added to the scope, even where competence in this general area has already been demonstrated.
Under flexible scope, laboratories are allowed to include additional activities in their scope of accreditation on the basis that their competence has been previously demonstrated to the Accreditation Body, not only to carry out activities according to previously evaluated procedures, but also for the development and validation of them following a pre-established system.
Recently, the Emergency Laboratory (EL) of the Department of Laboratory Medicine, Hospital Universitario La Paz, Madrid, Spain has renewed its biochemistry analyzers changing the Dimension RxL Max (RxL) to Dimension Vista1500 (Vista), both from Siemens Healthcare Diagnostics®. This implies some methodological improvements such as LOCI technology for the cardiac Troponin I, NT-proBNP and HCG measurements, C-reactive protein (CRP) by nephelometric assay and the possibility for calibration for enzyme quantification (5). As a result of the flexible scope accreditation and prior to use on patients, the laboratory always applies a validation strategy. This is based on CLSI recommendations to assess performance reliability and to grant the comparability of patient results (6-10).
The aim of this study is to show how to perform a validation of an analytical system, in this case a biochemistry analyzer, when it is introduced in a flexible scope accredited laboratory to guarantee their capacity of ensuring that the results are traceable for each individual patient.
Validation procedure involved evaluating the main analytical performance (imprecision and systematic error) of two Dimension Vista analysers and to assess the comparability of patient results of one of them with the previous methodology Dimension RxL.
Materials and methods
Study design
The Hospital Universitario La Paz is a tertiary institution containing four hospitals: the General Hospital, Traumatology and Orthopaedics, Obstetrics and Gynaecology and Children’s Hospital. It has nine critical care units, four emergency departments and some outpatient activities.
The EL receives analytical requests from all these hospitals. The laboratory has been accredited by the Spanish Accreditation Body (ENAC), according to ISO 15189:2007 since 2005. It was the first Spanish laboratory to obtain this accreditation and also the first to have a flexible scope accreditation. This accreditation covers all the analytes that are available in the EL.
A rigorous procedure established by the laboratory before the technological change was followed. The whole process was completed prior to use on patient samples. The initial step was to evaluate the analytical performance of two Dimension Vista analysers (namely V-1 and V-2). Secondly, a comparison was made between patients’ results obtained using Dimension Vista and using Dimension RxL Max. The analytical performance evaluation was made of the two Dimension Vista instruments and the comparison with Dimension RxL Max was made using only the results from one Dimension Vista (V-1).
The evaluation procedure had been based on the CLSI EP5-A2 document (8) for precision and CLSI EP-15 document (9) for bias estimation. CLSI EP9-A2 document (10) was followed for verification of comparability of patient results.
Multiple reagents and calibrator lots for all tests and only a single lot of control material were used over the study period. All materials were handled according to manufacturer recommendations.
Statistical analysis was performed in the evaluation software package StatisPro (CLSI) for the evaluation of imprecision, bias and total error at the medical decision levels based on tables provided by Westgard (11). Biological Variation (BV) was used as the main acceptance criteria (12) following the hierarchy established by the Stockholm consensus (13).
Evaluation of analytical performance of Dimension Vista 1500
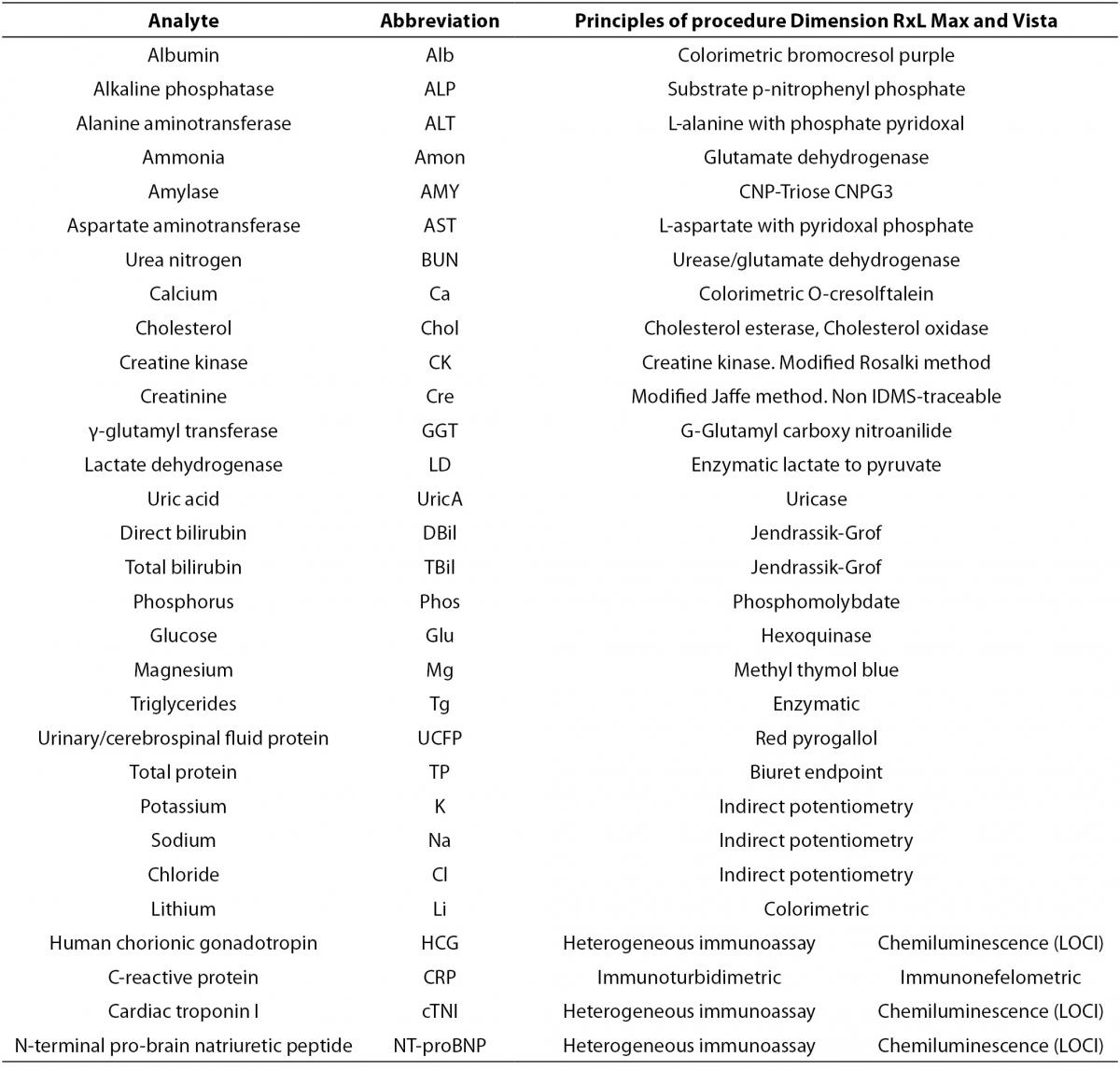
The scope of this study includes 30 analytes that were analyzed in the two Vista instruments and in the Dimension RxL Max. Principles of procedures of each parameter are shown in Table 1.
Table 1. Principles of procedures of Dimension RxL Max and Dimension Vista.
The selected test materials (Table 2) were reference materials from human serum or urine in order to be as similar as possible to the clinical samples. This quality control material was purchased from BioRad Laboratories Inc. (CA, USA). All of them were liquid, ready to use and they were stored and manipulated according to manufacturer recommendations. Vials which have been opened for more than two days were rejected.
Table 2. Quality control materials and pooled patient samples.
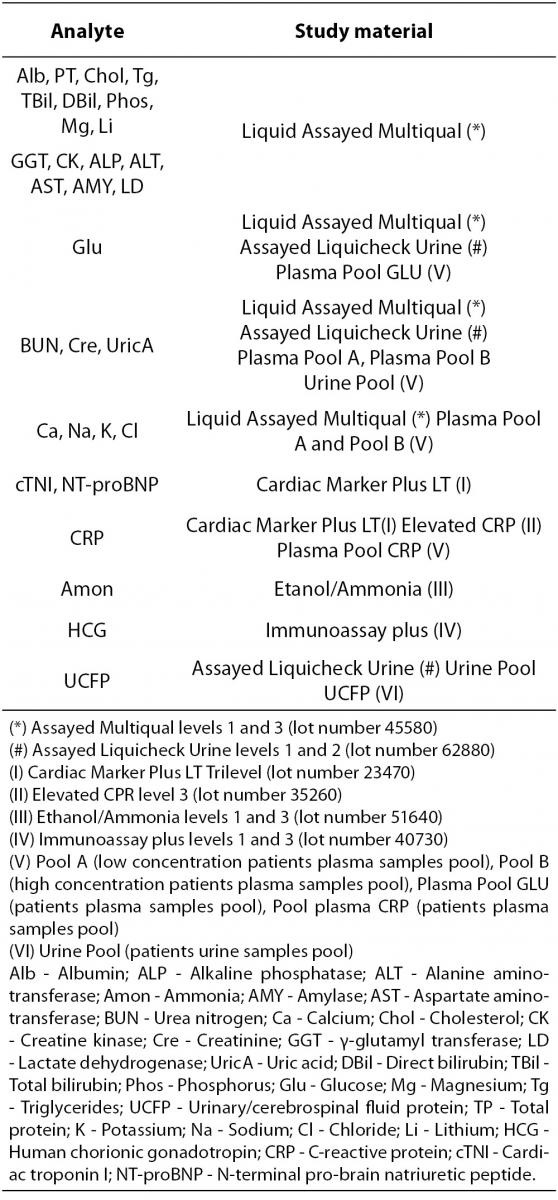
Pooled patient samples from plasma and urine were used to complete the study for some constituents. Their concentrations were set near medical decision levels. After centrifugation at 3500 rpm for 10 minutes all plasma and urine pools were separated, aliquots were made and frozen at -20 °C until analysis (5 days). Prior to use, following the laboratory procedure, they had been laid at room temperature until they had thawed out and homogenized before analysis.
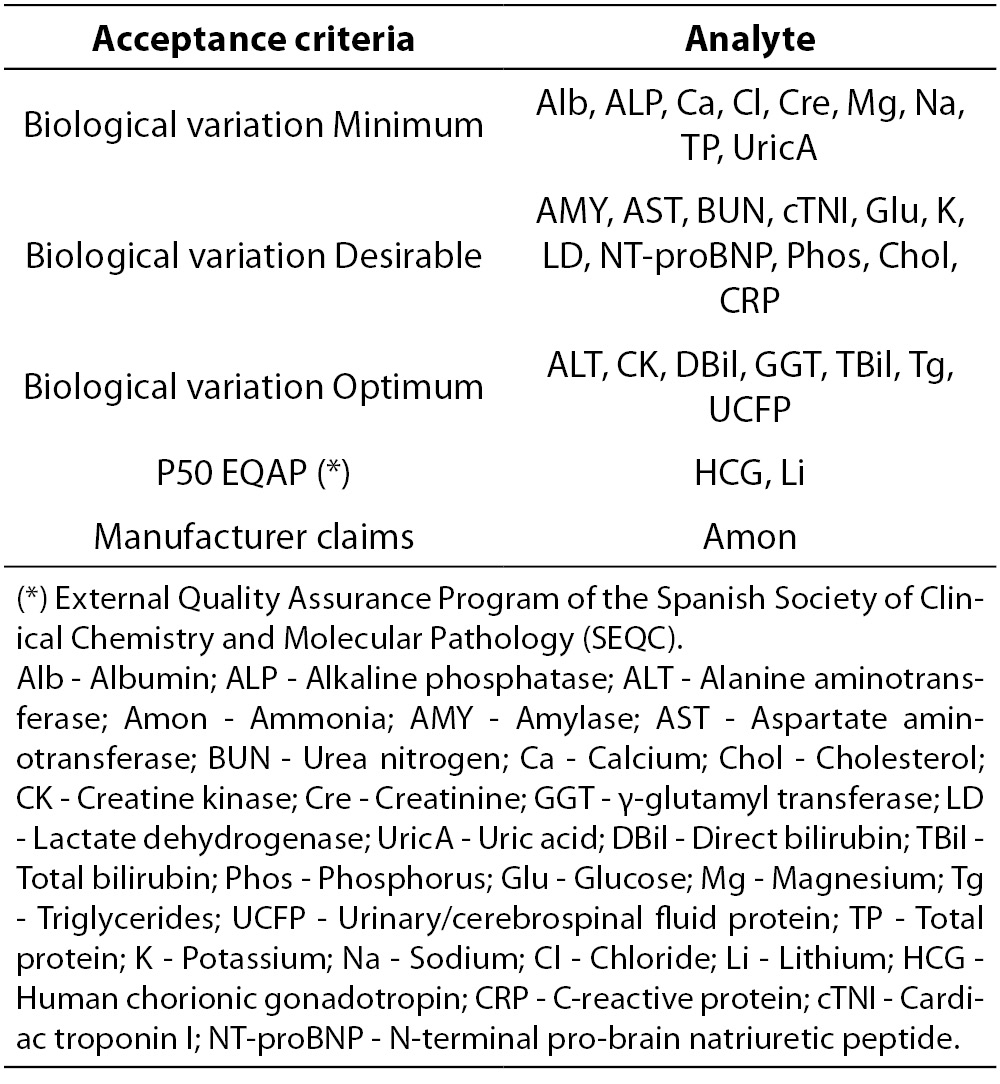
Laboratory acceptance criteria of analytical performance are shown in Table 3. Based on our former experience with Dimension RxL Max we assume the same criteria previously established as laboratory quality specifications. Imprecision and bias results should not exceed the respective acceptance criteria based on biological variation established by laboratory (13-15).
Table 3. Analytical quality acceptance criteria (imprecision, bias and total error) established by the laboratory.
Imprecision
After the preliminary familiarization phase (five days, 50 test per each parameter, 1500 patient analyses in total), the procedure was performed over a twenty day period. According to the recommendations of CLSI EP5-A2 document, the following steps were carried out per day: two runs of control samples material and pooled patient samples were analyzed daily. There was a maximum delay interval of two hours between the analyses in each device. Each run consisted of two replicates. Analysis was conducted by using a different aliquot per replicate. The run was discarded if there was any problem in quality control, procedures or any operating difficulty. Between each run the order of analysis of pooled patient samples and quality control material were interchanged.
To interpret the imprecision results different coefficients of variation (CV %) were calculated from all data (repeatability, between-run, between-day and within-laboratory imprecision). Results are expressed as within-laboratory imprecision per analyte and quality control material and it was compared with the allowable imprecision established by laboratory.
Systematic error or bias
Systematic error (SE) was estimated from experimental data obtained according to CLSI EP15-A2 document. Data were compiled upon every analyte and quality control level. Throughout this paper the term bias estimation is assumed to be equal to SE.
To interprete the bias results, bias was expressed as the difference between the average result that has been obtained for each parameter and the accepted reference value from a third party interlaboratory quality control program (Unity Real Time software, BioRad Lab) and it is expressed in terms of percent difference related to expected value (%). As accepted reference value, the cumulative media of the peer group from the third party quality control (BioRad Lab) was assumed (16).
Depending on the test, the peer group included 27-34 laboratories. The average number of the data included in the peer group was approximately 7000, though it varied from one parameter to another. The minimum data for any test was 5000, apart from urea nitrogen (N = 1988, provided by 13 laboratories).
The obtained values are compared with the allowable systematic error based on biological variation established by the laboratory as showed in Table 3.
Verification of comparability of patient results with the previous device
Forty randomly selected patient samples from the emergency laboratory, regardless of their clinical conditions were performed in two runs. Samples were analyzed in one of the Dimension Vista devices (V-1) and Dimension RxL on the same day of venous collection.
Analyses procedure followed the recommendations of document from CLSI (CLSI EP9-A2). Daily analysis of eight patient samples was undergone over a five day period. Two replicates of the samples were analyzed in the new device V-1. Later, within a 2 hour interval, the same samples were performed by the comparative procedure, also by duplicate.
To interpret the comparability verification results, the difference between the current method and the candidate replacement is considered as follows: Confidence interval of the estimated difference should include the laboratory acceptable error at the medical decision level. Total allowable error (TEa) also based on BV was considered as the criteria for comparison study.
Results
Tables 4 to 6 show the individual results of imprecision and bias evaluation of the two Dimension Vista analysers. They are grouped depending on the compliance degree of each parameter.
Table 4. Analytes that fulfil laboratory acceptance criteria for imprecision and bias in both analyzers.
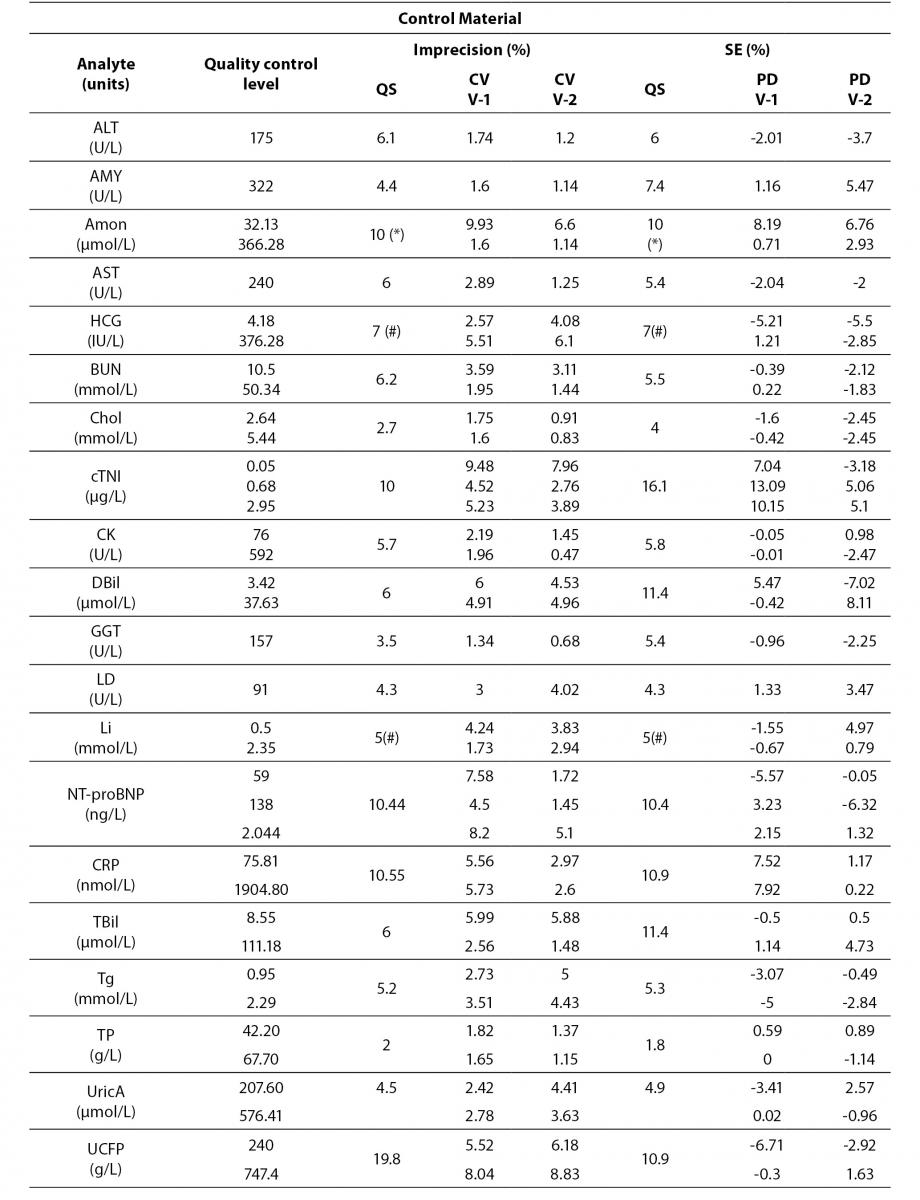
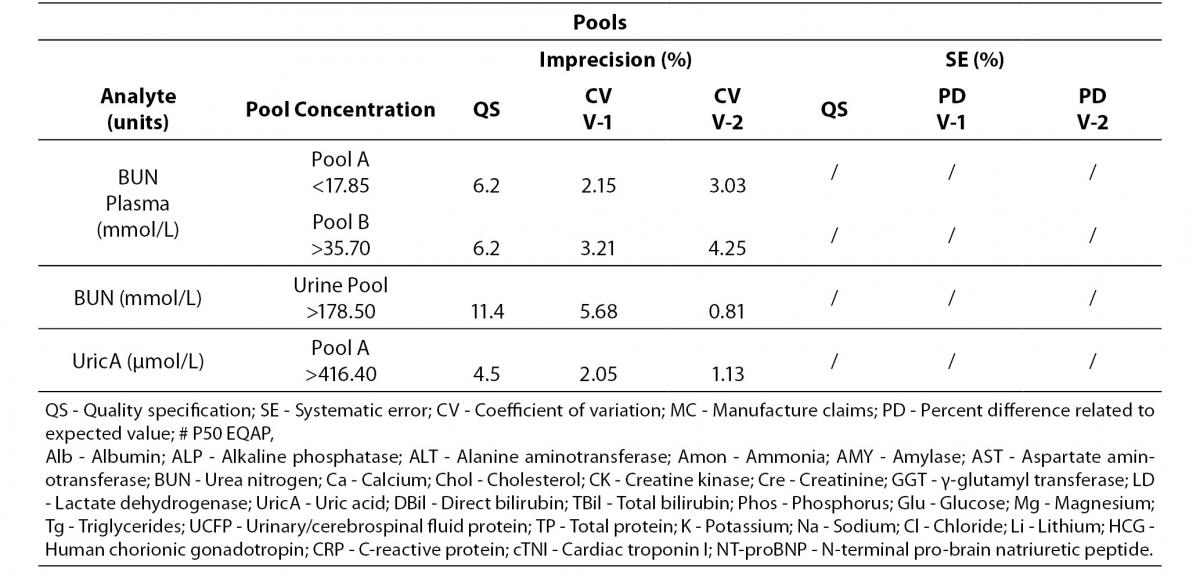
Table 4 represents all parameters which fulfil both imprecision and bias requirements in both analyzers. On the other hand, Table 5 highlights those parameters that do not reach the analytical requirement of imprecision or bias but they fulfil total error objectives (TEa = Bias + 1.65 x CV).
Table 5. Analytes within total allowable Error (TEa) in both analyzers.
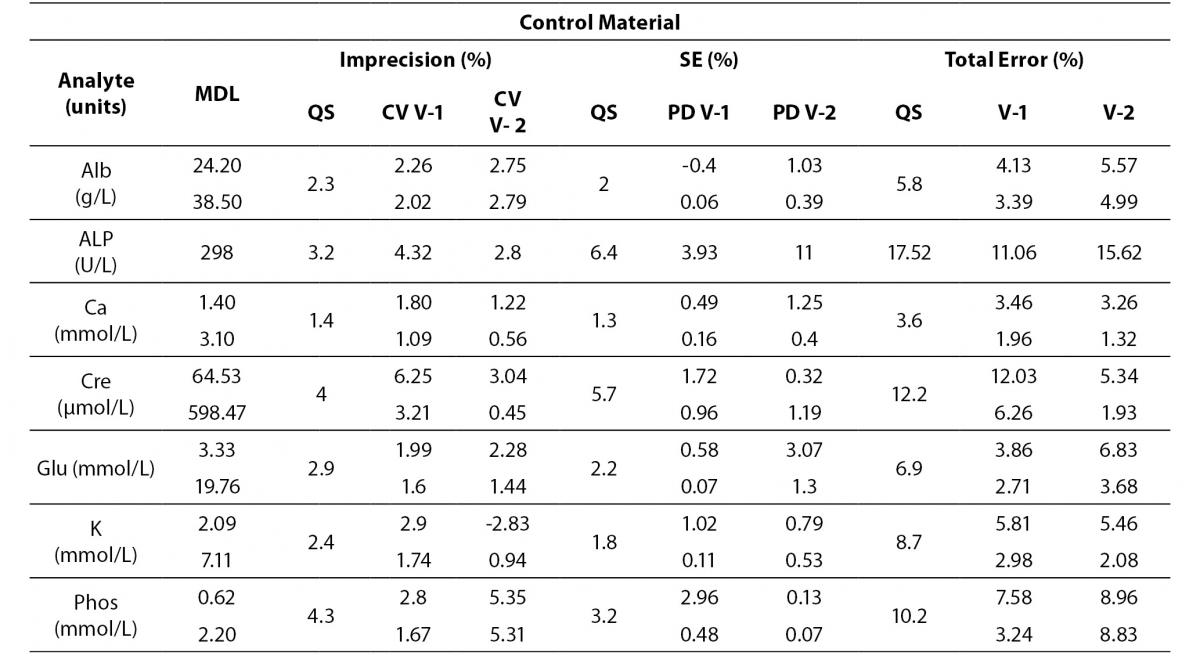
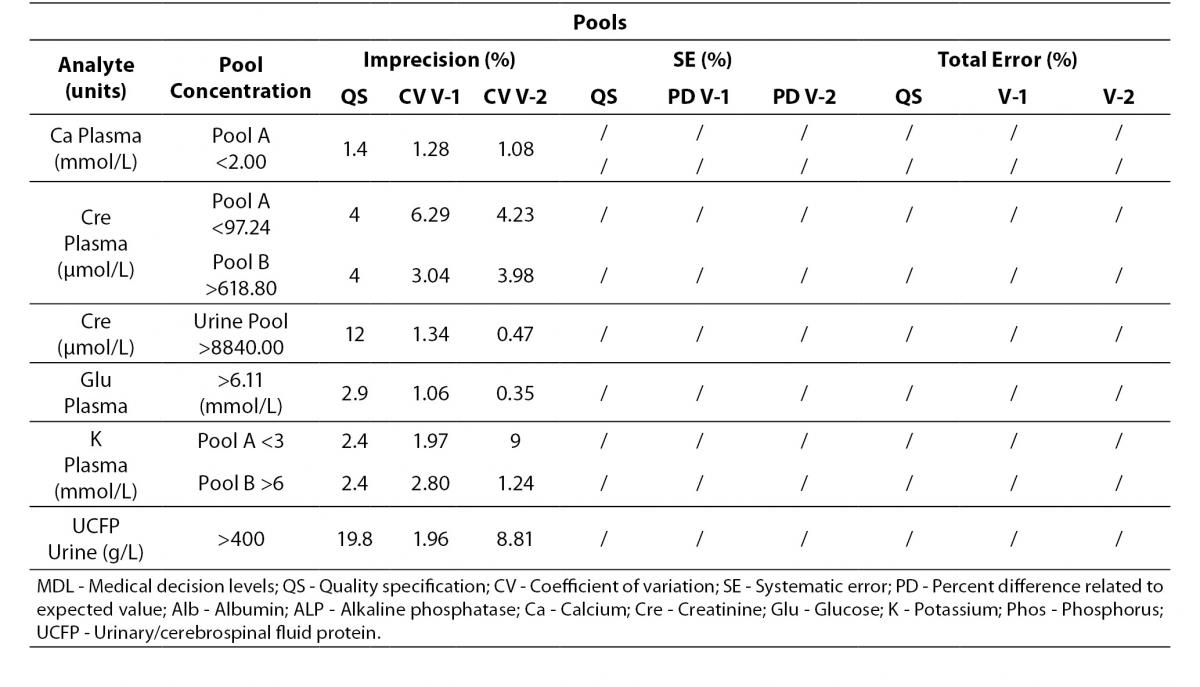
Analytes that did not fulfil any requirement are shown in Table 6. These analytes were considered to be in a special situation and were evaluated independently.
Table 6. Analytes that need further review in both analyzers.
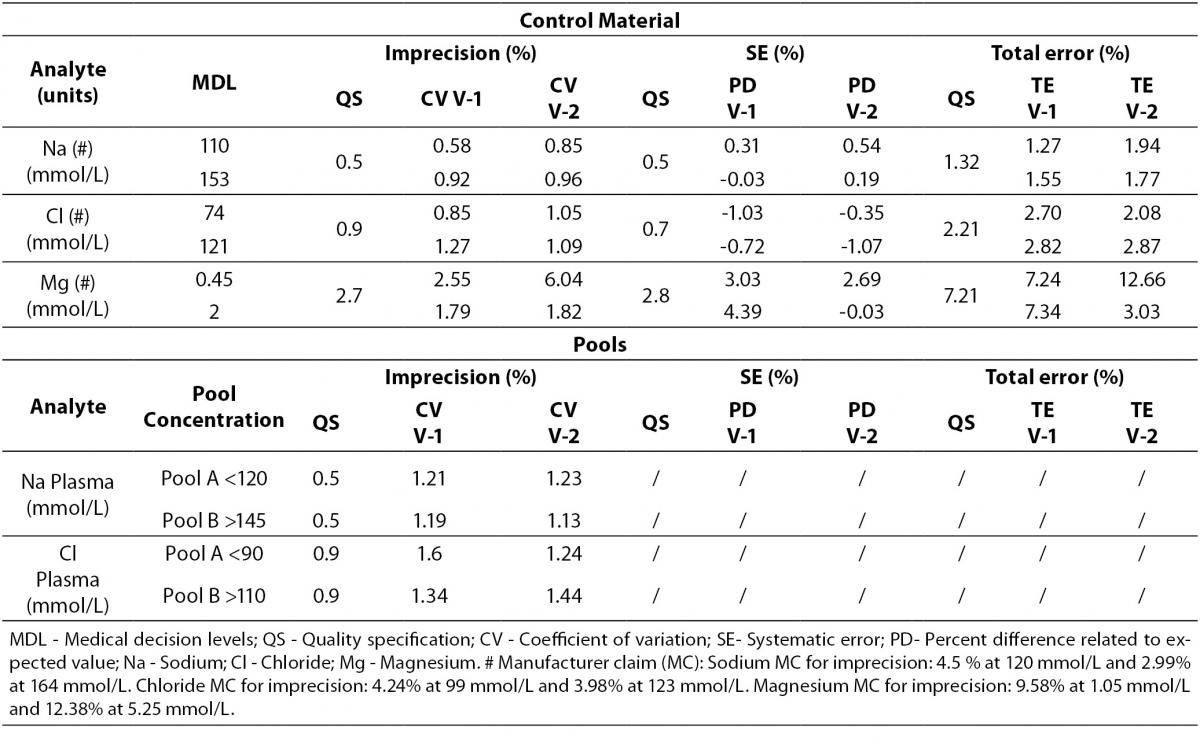
Regarding electrolytes specifically sodium and chloride, it is widely known that due to the current “state of the art” it is difficult to fulfil BV criteria (17). In fact, as a result of this evaluation, we do not reach the minimum imprecision BV criteria. Some actions were performed in order to achieve a better control of these parameters (such as cleaning system review, increasing maintenance protocols, water purification supplies review, etc).
Magnesium did not fulfil all the established analytical performance requirements. One of the devices had greater imprecision and the other showed greater bias than established by the laboratory as quality specifications. Therefore magnesium, as well as sodium and chloride, needed more actions to improve its performance. These included acting over preventive maintenance, improving calibrator stability by changing its storage conditions (frozen versus refrigerated) and even more, calibrator value was reassigned by the manufacturer.
Nevertheless, these three parameters (sodium, chloride and magnesium) fulfilled manufacturer claims (Table 6) and the Spanish minimum consensus quality specifications (17).
On the other hand, the verification of comparability of patient results is shown in Table 7.
Table 7. Verification of comparability of patients results with the previous device.
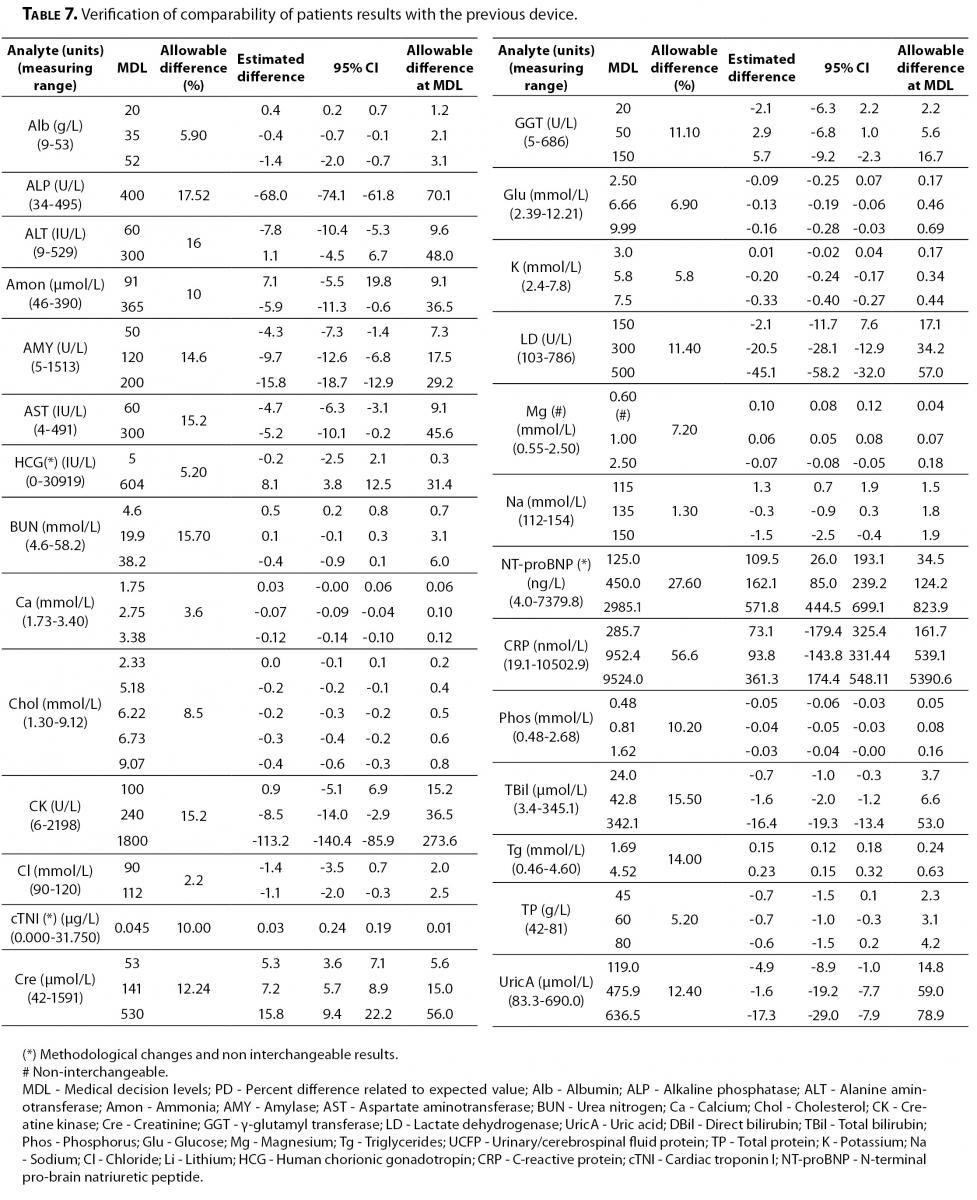
All the parameters performed by the same methodology were interchangeable with the previous device, except magnesium in which significant differences were found. Analytical conditions and reference intervals were reviewed, showing no additional problems. Finally this issue was solved as the manufacturer changed the calibrator assigned value after conducting an interim review process. The change was intended to better align the Dimension Vista magnesium results with the reference method, atomic absorption, particularly at the low end of the assay range (Siemens HD Support Bulletin DV-1034_MG Dimension Vista® CHEM 1 Calibrator).
The parameters with methodological changes such as cTNI, NT-proBNP and CRP obviously showed that patient results were not interchangeable.
Discussion
Patients belonging to a healthcare system, at some stage within their life, may encounter different systems of measurements within the same institution. This may be the case even during a single clinical event. It forces the laboratory, as a critical point, to control and ensure that all the results that may be issued are comparable (18). In addition to this, the laboratory must ensure that they are incorporated into each patient’s medical record, without distorting or creating errors which may affect the interpretation of their results (19) and CLSI recommends to control this aspect as a means of risk management (6,7).
In addition, this represents an ISO 15189:2007 requirement and accreditation bodies must review that laboratory had performed all the necessary mechanisms to ensure this issue.
The flexible scope in the accreditation process (2-4) implies that laboratory assume responsibility for the management of all or part of its scope of accreditation without the necessity of a preliminary evaluation by the Accreditation Body for each new activity. For which reason, the laboratory must demonstrate to the accreditation body that it has the technical competence in evaluating methods prior to use on patient samples. As an accredited laboratory this enables it to incorporate a new test into its accredited scope on the basis of its own validations without waiting for the evaluation by the accreditation body prior to the next audit or having to undergo an extraordinary audit.
The strength of the flexible scopes of accreditation is based on the capability of the laboratory’s management system on the implementation of the validation and/or verification procedures and the monitoring activities related to their implementation (appropriateness of claims, personnel competence, estimations of measurement uncertainty, equipment and measurement traceability, proficiency testing activities and internal quality control).
In this context accreditation body evaluation is focused on laboratory validation activities. That implies that the laboratory has to develop a well organized and documented procedure of validation of the measurement system so that it could be able to demonstrate the adequacy of the system in an easy and reproducible way. This is very important in order to shorten the time of evaluation and reducing the impact of the economical and human resources expenses, increasing the laboratories’ effectiveness.
On the other hand, the main objective of the laboratory is to serve as a diagnostic tool as well as monitoring a treatment or a disease evolution. In this context, it is necessary to ensure that the analytical quality of the results fulfil their clinical needs. Our laboratory established biological variation as the criteria which enable this (12).
Nevertheless, all procedures to guarantee analytical quality should be performed in a standardized way. For every kind of evaluation our studies follow the CLSI protocols, recognized worldwide. This includes EP5 for imprecision, EP15 for bias estimation and EP9 for method comparison.
One of the remarks that CLSI highlights in these studies is the kind of selected material. It is recommended to be as similar as possible to patient samples. The other remarkable need is the concentration at which clinical decision is made. They should be as near as possible to the medical decision levels. Samples selected in our studies are quality control material from human matrix and they are complemented with patient samples pools from plasma or urine for some parameters of interest. All comparison studies were performed on patient samples.
The medical literature deals with these kinds of issues by approaching only a few parameters at one time (20-22). Other studies only deal with a single analytical performance evaluation (23,24). There are not many articles that include all routine biochemistry parameters in the same procedure (25,26) covering the main evaluation protocols. On the other hand, the majority of studies set the manufacturer claims as quality objectives (25-27). Biological variation which constitutes the analytical objectives of our study is generally more stringent than manufacturer claims (28).
Related to the evaluation of analytical performance of Dimension Vista, 27 of 30 studied parameters fulfilled all analytical criteria. Even though 7 of them did not reach the desired imprecision or bias, all were less than TEa, therefore all were considered within acceptance criteria.
If an analyte shows results greater than the allowable imprecision or bias, but are less than the total allowable error (the maximum acceptability limit for laboratory), it could be considered acceptable.
The 3 parameters that did not fulfil the analytical requirements (sodium, chloride and magnesium) needed further evaluations and actions before being used on clinical patients. All fulfilled manufacturer claims and also the minimum consensus analytical specifications derived by the “state of the art” that has been published, as well as other well known specifications (17,29-31). As previously described, these actions included changes in maintenance protocols, review of water supply conditions, changes in storage conditions of calibrators and reassignation of calibrator values.
Due to the thorough nature of the study, we were able to highlight some analytes with poor analytical performance that otherwise could be overlooked. This is one of the main benefits from the study since we could apply a more stringent quality control strategy over these parameters, having a positive impact on clinical practice.
On the other hand, this study considers the use of biological variation criteria as an analytical quality objective instead of using other wider objectives. Depending on the laboratory resources and the “state of the art”, some of these quality specifications might be unrealistic. If the actual objective is not achievable in a long term monitoring, after acting in a corrective way, it is important to consider the possibility of changing the quality objective. In that case, if the chosen objective is down in the Stockholm hierarchy, the laboratory must assume that the level of quality necessary to satisfy medical needs may not be fulfilled.
This was not our case because TEa biological variation specifications were mostly reached, as was confirmed in the monthly laboratory periodic evaluations.
All parameters included in the new technological improvement (LOCI) (32) obtained better analytical performance than previous methodologies: cTNI, NT-proBNP as well as CRP by nephelometrical assay.
Regarding these parameters, some changes have been made. For instance, a new 99 Percentile cut-off for cTNI was established (0.04 ng/mL) highlighting that imprecision less than 10% can be obtained for this cut-off (33,34). Clinicians were alerted about this new diagnostic point by means of multidisciplinary clinical seminars, written warnings, etc.
Regarding the verification of comparability of patient results with the previous device, all parameters performed by similar methodologies were interchangeable, except magnesium.
In the case of magnesium, significant differences were found at the medical decision levels for hypomagnesaemia diagnosis which is relevant in children with kidney transplant for instance. Clinicians were alerted about the possibility of new reference population values and advised that every individual patient should have been carefully analyzed and compared with their historical record. Finally, the problem was solved with the new calibrator assigned value by the manufacturer.
These evaluations illustrate that even same methodology and same manufacturer can give non-interchangeable patient results. In addition, a different device of the same model can produce different analytical performance.
In summary, a new device installation should always be validated by the laboratory due to the fact that different working conditions can influence patient results. This kind of evaluation procedure should always be performed prior to use on patient samples. Therefore, laboratory can demonstrate some limitations that otherwise could be overlooked.
Dimension Vista showed a generally good acceptable analytical performance according to biological variation criteria but technical supervising should be necessary for some parameters such as sodium or chloride. Some other parameters met biological variation criteria except at the medical decision levels (creatinine, magnesium) and would require the application of extreme quality control procedures.
The benefit of having a flexible scope of accreditation is to be able to include additional or new activities as accredited from the first patient result. In that way it is not necessary to be previously evaluated by the Accreditation Body. Laboratories having flexible scopes can reduce the economical impact of technical innovations increasing their laboratory effectiveness.
Acknowledgments
We thank Siemens Healthcare Diagnostics and BioRad Laboratories Inc, for reagents and reference materials supplied for this study.