Introduction
Endothelins were first identified in a paper by Yanagisawa et al. as potent vasoactive peptides, regulating vascular tone, blood pressure, cell proliferation and hormone production (1). Diversity in biological function became rapidly apparent with many other roles for these peptides now described (2,3). To date, three isoforms of endothelins (ET-1, ET-2 and ET-3) coded by three different genes have been identified (2). The human gene for ET-1 is located on chromosome 6, the ET-2 gene is on the first chromosome, and ET-3 gene is on chromosome 20 (2). The plasma concentration of ET-1 in adults is normally between 0.4–8.1 pg/mL (4). Each isoform, however, shows a tissue type-dependent pattern of expression. ET-1 is widely expressed in endothelial cells, vascular smooth muscles, central nervous system (CNS), and reproductive tissues. Levels of ET-2 are high in the intestine and kidney, whereas ET-3 is mainly expressed in the brain (5,6).
Endothelin biosynthesis starts from the precursor molecule called pre-pro-endothelin (pre-pro-ET), which in humans consists of 212 amino acids. Pre-pro-ET hydrolyzes to “big ET” or pro-endothelin (pro-ET). Furthermore, endothelin converting enzymes (ECE) in the secretion of pro-endothelin converts pro-endothelin to mature endothelin (ET).
Endothelin converting enzymes (ECE)
Endothelin converting enzymes are atypical endopeptidases (metalloproteases), predominantly found in the same, or in close proximity to, those cells expressing each endothelin (7,8). Three forms of ECE have been described: ECE-1 (9), ECE-2 (10), and ECE-3 (11), with different specificities for the isoforms of big ET. ECE-1 specifically hydrolyzes the Trp21-Val/Ile22 bonds of big-ETs to produce biologically active ETs (12,13). Four isoforms of human ECE-1 (1a, 1b, 1c and 1d) have been identified to date (12,14). The four proteins are encoded by one gene, but each is expressed from a distinct promoter that regulates the expression of the four unique amino termini (12). Similarly to ECE-1, four different isoforms of ECE-2 have been identified: ECE-2a-1, ECE-2a-b, ECE-2b-1 and ECE-2b-2 (15), also with different subcellular localizations (16). ECE-3 is specific for big ET-3 (11). Previous studies indicate that ECE-1 is the main enzyme responsible for the transformation of big ET into ET (16). Several studies have shown that ECE-1 efficiently hydrolyzes a number of peptide hormones other than ETs, these include bradykinin, substance P and neurotensin (12,14). An exciting novel substrate for ECE-1 is the beta-amyloid peptide that is implicated in the pathogenesis of Alzheimer disease (12). Inhibitors of ECE-1 are considered to be valuable therapeutic agents and were developed for the treatment of various disorders linked to elevated ET-1 levels (12,14). Numerous peptides or non-peptidyl ECE-1 inhibitors have already been produced, but neither is currently used for therapeutic purposes, perhaps because of insufficient knowledge of the ECE-1 family of proteins in naturally expressing cells.
Factors affecting the secretion of endothelin
ET-1 is the most potent vasoconstrictor in the ET family, exhibiting a long duration of action. This peptide is produced by vascular endothelial cells and is the major form of ET in the circulation. Stimuli for ET synthesis are the mediators which can be divided into three groups:
- Local factors (activated platelets, endotoxins, thrombin, cytokines (IL-1), growth factors, oxidized LDL);
- Hormones (antidiuretic hormone, adrenaline, insulin, angiotensin II); and
- Physical factors (hypoxia, hypovolemia, slightly tangential strain the arteries) (17-21).
Inhibitors of endothelin synthesis are: nitric oxide (NO), atrial natriuretic peptide (ANP), prostaglandin E2 (PGE2), prostaglandin E1 (PGE1), heparin, hepatocyte growth factor (HGF), epidermal growth factor (EGF) and strong shear stress (increased blood flow) (17,22). Recent studies have shown that the generated ET-1 may be stored in endothelial cells, which are the main biological source of ET-1 (17). Normal plasma level of ET-1 is low (0.4–8.1 pg/mL), which is insufficient to activate endothelin receptors. It is believed that the ET-1 levels are higher in the intercellular space between endothelial cells and vascular smooth muscle cells (4,17).
Endothelins are secreted in two ways: the constitutive and the regulatory. Constitutive secretion is the most common type of supervision and involves gene transcription and increased stability of mRNA for pre-pro-ET. In this way the secretion after appropriate stimulus is very slow. In the regulatory mode, stimulation leads to rapid secretion of ET. ET-1 half-life is less than 5 minutes, yet it manifests its effect much longer (23) (Figure 1.).
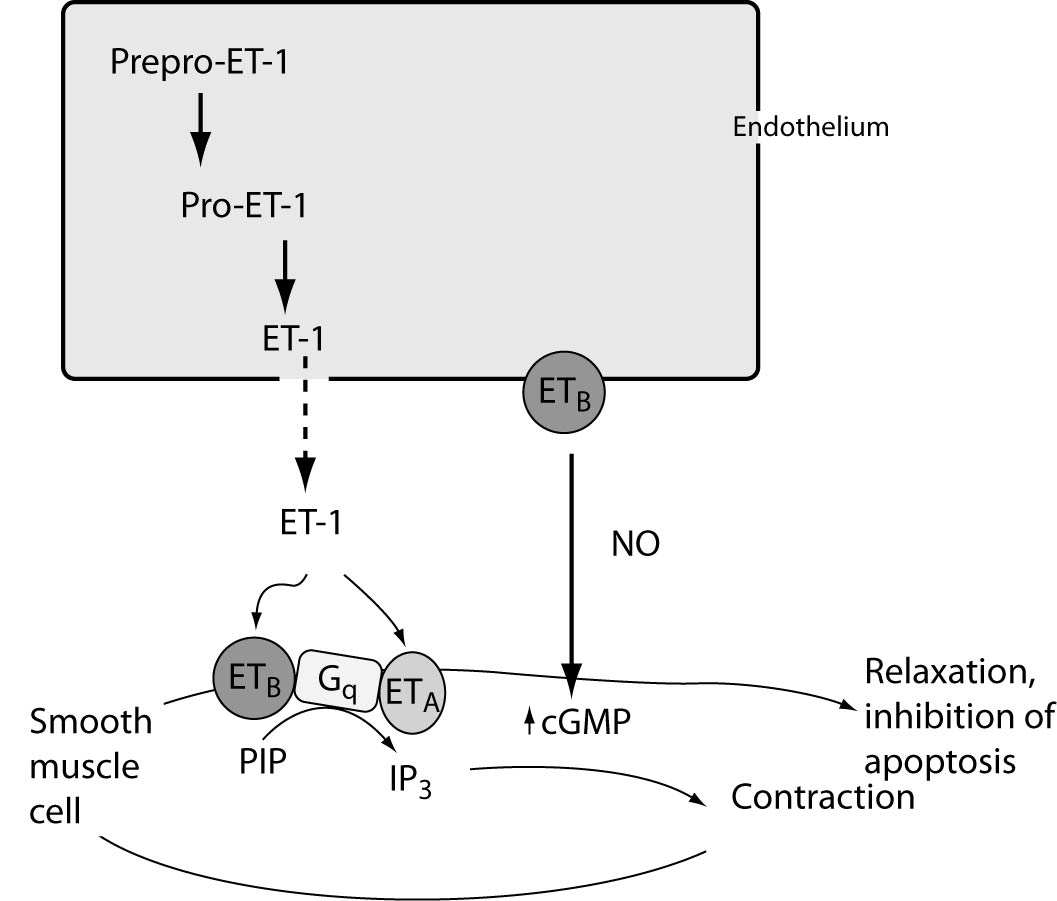
Figure 1. Regulation of ET-1 synthesis, pathway of ET generation and ET-receptor-mediated actions on smooth muscle cells. The product of ET1 transcription is prepro-ET-1, which is cleaved by a neutral endopeptidase to form the active precursor pro-ET-1 or big ET-1. Big ET-1 is converted to the mature peptide by the metalloproteinase endothelin-converting enzyme-1 (ECE-1). Two ET receptors have been identified in the vasculature: ET type-A receptors (ETA) reside in vascular smooth muscle cells and mediate vasoconstriction and cell proliferation, whereas ETB receptors reside on endothelial cells and are mainly vasodilatory through NO (which in turn can mediate the anti-apoptotic effects of ET-1), and regulate the synthesis of ET-1. ETB receptors on smooth muscle cells can elicit vessel contraction.
Endothelin receptors
The biological effects of ET production are determined via activation of one of two G-protein coupled receptors, endothelin receptors A (ETRA) and B (ETRB1 and ETRB2). Recently endothelin receptor C (ETRC) was discovered, but its´ functions and distribution are still unclear (24). Endothelin receptors subtypes are also distinctive in their localization, with colocalization to various tissues signifying the presence of autocrine and paracrine functions. ETRA can be found in many human tissues (vascular smooth muscle, heart, lung and kidney) while expression in the endothelium does not exist. ETRB is primary expressed in the brain (cerebral cortex and cerebellum), moderate in the aorta, heart, lung, kidney and adrenal gland. Recently, subtypes of ETRB have been described (25). ETRB is highly expressed in the endothelium (ET (B1)), as well as in vascular smooth muscle (ET(B2)). Selectivity of these receptors for certain isoforms of endothelin was also proved. ETRA has a high affinity for both ET-1 and ET-2 (and low affinity for ET-3) whereas ETRB has a similar affinity for all the isoforms (25). Upon activation by ligand binding, ETRA and ETRB often elict opposite physiological responses in their target tissues. Activation of ETRA leads to vasoconstriction, bronchoconstriction and secretion of aldosterone. Activation of ETRB1 stimulates the formation of NO, PGI2 and endothelium-derived hyperpolarizing factor (EDHF) and causes vasodilatation. In contrast, activation of ETRB2 receptor causes vasoconstrictive response. It is still unclear whether the various biological effects of endothelin are mediated by activation of different signaling pathways, which probably contribute to different responses to stimuli. Binding of ET-1 to cell surface of ETR results in the generation of a number of second messengers; these messengers activate kinases or phosphatases, which in turn phosphorylate or dephosphorylate downstream signaling molecules, leading to a diverse range of biological responses. ET-stimulated effects can be classified into short-term and long-term responses, which are regulated trough different signaling pathways. In short term responses, ET-induced phospholipase C (PLC) activation leads to the Ca2+ generation of inositol-1,4,5-triphosphatase (IP3) resulting in influx, and of diacylglycerol (DAG), resulting in protein kinase C (PKC) activation. ET-1 stimulates the translocation of cytosolic PKC to the plasma membrane, and PKC subsequently phosphorylates specific cellular proteins, resulting in multiple biological effects (ET-> ETR-> PLC-> PKC). In some tissues ET-1 also activates phospholipase A2 (PLA2) and leads to the formation of arachidonic acid metabolites, including leukotrienes, prostaglandins, and thromboxanes (ET-> ETR-> PLA2). In long-term responses, ET-1 plays a significant role in the chronic modulation of cell proliferation and differentiation, and MAPK (mitogen-activated protein kinase) may serve as the key regulator in these nuclear and cytoplasmic events (25-27).
Effects of endothelin on organs function
Endothelin gains its effects mainly with paracrine and in minor part with autocrine mode of action, and that is supported by the fact that the ETRs were usually placed on or near the membranes of the cell which produces ET (28). Studies with endothelins and specific endothelin-receptor antagonists have suggested that these peptides are important in vascular physiology (29).
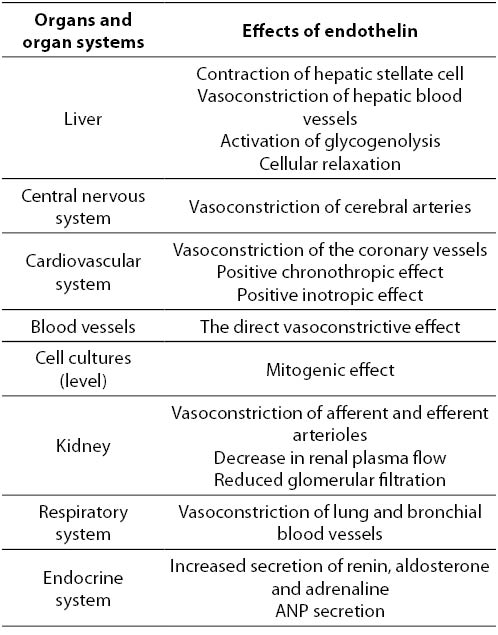
Effects of ET-1 on organs and organ systems can be divided, as mentioned previously, into short-term (hormone secretion, muscle contraction, control of cellular transport) and long-term (control of gene expression, effect on cell hypertrophy, mitogenesis, etc.). ET is a paracrine mediator, which stimulates the release of several hormones including ANP, aldosterone, adrenaline and hypothalamus and pituitary hormones. High concentration of ET-1 was observed in thyroid follicles and in the amniotic fluid, and therefore it could be considered that the ET-1 participates in the synthesis of thyroglobulin and in controlling of uteroplacentary blood flow (28). Also, ET-1 causes renal and cerebral vasospasm, and has an important role in the development of cardiorespiratory syndrome (28). Strong experimental proofs indicate the role of ET in the pathophysiology of certain disorders such as myocardial infarction, coronary vasospasm, acute renal failure, Raynaud’s disease, asthma, primary pulmonary hypertension and fibrosis in liver disease (2,3). The role of ET in essential and renal hypertension, and in left ventricular hypertrophy is not well supported experimentally (28) (Table 1).
Table 1. Effects of endothelin on the organs and organ systems
Role of endothelin on liver function
Distribution of ETRs on the membranes of liver cells varies. A diversified presence of ETRB has been confirmed on endothelial cells of sinusoidal capillaries, hepatic stellate cells (HSC – Hepatic stellate cells of fat store), portal vein endothelial cells and epithelial cells of the bile ducts. Previous studies have shown the highest expression of ETRA and ETRB on the membranes of HSC cells and on the membranes of hepatocytes (30,31). ET-1 is synthesized in endothelial cells of sinusoidal capillaries, HSC cells and in epithelial cells of bile ducts. Several studies reported a weak expression of ET-1 in the healthy liver cells, while in liver cirrhosis significantly higher synthesis of ET-1 was noted. These findings were supported by the increased expression of mRNA for ET-1 isolated from HSC in the cirrhotic rat liver biopsy (32,33). Several studies have shown that ET-1 stimulates the reversible contraction of HSC in culture, mediated by elevated intracellular calcium via the both ETRA and ETRB (32-34). Elevation of intracellular calcium and subsequent HSC contractility was reduced after administration of BQ-123 antagonist of ETRA. Thus, it can be concluded that the HSC contractility is probably mediated by ETRA and that in chronic liver disease ET-1 regulates the flow through the sinusoids by stimulating contraction of HSC (32). There is also evidence that the release of ET-1 promotes vasoconstriction of the hepatic veins (35). Furthermore, it is confirmed that high concentrations of ET-1 reduce bile flow, which probably plays a role in the pathogenesis of cholestatic liver disease. Coupling of ET-1 with the ETRB on the membrane of hepatocytes activates system of phospholipase C leading to the release of calcium from the intracellular pool and blocks the efflux of calcium from the cells which results in long lasting and strong activation of glycogenolysis (36). Binding of ET to receptors in the liver do not have only vasoconstrictive effects. Binding of ET-1 to ETRBs on the cell membrane of Kupffer cells activates PLA2, which leads to the release of prostaglandin E2 and NO that mediate vasodilation (37,38).
The role of ETRA and NADPH oxidase in the vascular abnormalities
Normal vascular activity is essential for maintaining normal function of organs depending on the balance of vasoconstrictive and vasodilative substances derived from the vascular endothelium. Substances mainly include NO to dilate and ET-1 to constrict the vascular smooth muscle. Endothelium-dependent relaxation directly relates to the biosynthesis and release of NO by the activity of endothelial NO synthase (eNOS) (39,40). Endothelial dysfunction characterized by impaired endothelium-dependent vasodilatation has been linked to each of the known risk conditions (diabetes mellitus, hypertension, dyslipidemia, obesity, smoking and aging). The more impaired vascular endothelium, the more severe is the risk for cardiovascular diseases, thus it is likely to take an impaired vascular endothelium as the early stage of coronary infarction (39). Increased activity of NADPH oxidase (NOX) in diabetes mellitus leads to increased levels of reactive oxygen compounds (ROS). It has been widely accepted that ROS induced by hiperglycemia contribute to vascular endothelium dysfunction in diabetes (27,39). Concentrations of ET-1 are also elevated in patients with diabetes mellitus, expression of ETRs in blood vessels is increased and bioavailability of NO is reduced. NOX is stimulated mostly by ETRA via PKC activity (39). The vasoconstrictive activity of ET-1 is mediated by the activity of NOX, and an activation of L-type calcium channels by ET-1 causes an influx of Ca2+ through activating NOX. An interaction of ET-1 with ROS is known as ET-ROS signaling pathway, which activates ERK signaling pathway and AP-1 transcription factor (41). The impaired vasodilative responses and the reduced NO bioavailability may be addressed by an excess of ET-1 resulting in up-regulation of ETRA in the vascular wall, due to diabetes, hypercholesterolemia, hypertension and aging (41). Agents reducing hyperglycemia and hypercholesterolemia may not be sufficient in relieving the endothelial insults. A reversal of the abnormal endothelial status can be achieved by cell protective effects through suppressing the ET-NOX-PKC pathway. Agents may relieve the endothelium dysfunction by suppressing this pathway. Use of medications that suppress activated ET-NADPH oxidase in the vascular wall may diminish vascular abnormalities. Testing of the activity of these agents is valuable for the further potential in relieving abnormal vascular activity, reducing the risk of morbidity and mortality in patients at risk (39).
Role of endothelin in the pathophysiology of cerebral vasospasm
Recent studies have shown that changes of the ETR expression and function in the wall of cerebral arteries are a considerable factor for development of cerebral vasospasm (CVS) (42).
Under physiological conditions, the dominant vasocontractive effect of ET-1 is mediated by ETRA on smooth muscle cells (SMC), which is attenuated by an ETRB dependent release of NO from endothelial cells (EC). In the physiological cerebrovasculature ECs express exclusively ETRB and SMCs only ETRA. In the case of CVS an increased expression of the ETRB could be detected in cerebral vessels. However, the loss of the vasodilative and the missing of a vasocontractile ETRB mediated effect was demonstrated (42). Therefore, any ETRB mediated vasoactivity seems to be lost in the case of CVS and the biological impact of the increased expression remains unclear so far. The ETRA expression seems not to be increased during the development of CVS (42). Therefore, the proven increase of the ET-dependent vasocontractility seems to be the result of deficiency of ETRB mediated effect rather than the result of increased ETRA activity. Despite of the more significant changes of the ETRB expression, the pathophysiological effect of ET, namely the vasoconstriction, seems to be exclusively mediated by the ETRA (42). Therefore, tailored approaches for the treatment of CVS remain to be ETRA receptor selective antagonists.
Effects of endothelin on smooth muscle cells and cardiomyocites
It was believed that smooth muscle cells expresses only ETRA. More recently, ETRBs have also been identified in vascular smooth muscle cells, and these two receptors mediate vasoconstriction (43). The presence of ETRB with the variation of ETRA/ETRB ratio was detected on endothelial cells and it is higher in arteries than in veins. Interestingly, the level of ETRB expression in smooth muscle cells can also be increased in vascular disease (44). The main role of ET is smooth muscle cells-mediated vasoconstriction. However, the precise role of ET-1 and ETRA in the maintenance of systemic circulation and their contribution to the maintenance of blood pressure is not clearly understood. Unlike the ETRB which is explored by genetic models, knowledge about the impact of ETRA in the regulation of blood pressure arises only from studies of pharmacological antagonists (45). Model of intentionally induced hypertension has shown that antagonists of ETRA suppress increase or even partially reduce mean arterial pressure after induced hypertension (45). Therefore, it seems that antagonists of ET-1 receptors may play an important role in treating some forms of hypertension. However, in spite of their antihypertensive effect, the usage of ETRA antagonists remains a problem, mainly because of potential side effects and the availability of safer drugs such as ACE inhibitors (angiotensin-converting enzyme inhibitors) or antagonists of angiotensin II.
Effects of endothelin in pulmonary arterial hypertension
Pulmonary arterial hypertension (PAH) is only condition when ET receptor antagonists are effective as antihypertensive drugs (44,45). Various studies have shown that ET receptor antagonists prevent the development of PAH, both pulmonary vasoconstriction and hypertension (44-46). It is still unclear why the pulmonary circulation is especially sensitive to such ET-1 regulation. Therefore, the lungs remain the target organ for testing antihypertensive effect of ET receptor antagonists (47,48). There are two commercially available ET receptor antagonists for the treatment of PAH: ambrisentan and bosentan. Significant improvement was described in symptoms of patients treated with these drugs (48). Coronary endothelial cells are a major source of ET-1. On cardiomyocites ETRA and ETRB were found, with predominant ETRA. ET-1 causes a positive inotropic effect on cardiac muscle cells. It was also observed that ET-1 affects the contractile ability of myocytes (48). Binding of ET-1 on the ETRA stimulates the formation of IP3 and diacylglycerol by PLC and activation of PKC (ET-1-> ETRA-> PLC-> PKC). Activated PKC stimulates Na+/H+ exchange. Increased contractile effect of myocytes is a result of calcium release from the endoplasmic reticulum (ER). The sensitivity of cardiac myofilaments on endothelin binding leads to alkalosis mediated by Na+/H+ exchange and increased release of calcium leads to the accumulation of Na+, mediated by Na+/H+ pump (49). It has been shown that ET-1 causes hypertrophy in culture of neonatal rat cardiac myocytes, and it is considered that it leads to hypertrophy of adult cells. Recent studies have shown that transcription factor ZFP 260 reduces blocked ET-1 induced hypertrophy of cardiomyocytes (49,50). In other words, it was shown that increased expression of that transcription factors was involved in the induction of hypertrophic response (50).
Effects of endothelin on fibroblasts
Fibroblasts are located in the extracellular matrix (ECM) of connective tissue, where they serve as the main helpers of ECM synthesis, not only in the cardiovascular system, but in the whole organism. Fibroblasts are normally present in tissues, but they are also able to modify the phenotype (myofibroblasts) during the regeneration of tissues. During normal wound healing, myofibroblasts disappear and organ function is restored. However, in abnormal wound healing they are withheld in locations of tissue damage which leads to delay hypercontractile ECM, which is the main characteristic of fibrotic conditions such as idiopathic pulmonary fibrosis (IPF) and scleroderma (SSc) (51,52). Fibroblasts synthesize ET-1. Some studies have demonstrated increased levels of ET-1 in the fibrotic conditions (after lung biopsy in scleroderma) (52,53). In recent years, ET-1 is considered as a profibrotic factor and it’s suitable for research in the treatment of fibrotic conditions. It has been already proven that bosentan (ET receptor antagonist in PAH) prevents the formation of finger ulcers in patients with scleroderma (54). Numerous studies have described the initial phase of the development of fibrosis which involves the injury of endothelial cells of blood vessels when the release of mediators of inflammation occurs (NO and ET-1) (53,54). These factors are mitogenic for endothelial cells surrounding the injury. Therefore, it is believed that the injury of blood vessel presents pathological mechanism that leads to tissue fibrosis. The presence of such vasculopathy is a feature of SSc, which is clinically manifested as Raynaud’s Syndrome and ulceration of the fingers (55). Whether ET-1 is key mediator of vasculopathy has not yet been fully clarified. So far, studies have shown a correlation between Raynaud’s Syndrome and plasma ET-1 concentrations in patients with SSc. This thesis is supported by clinical use of bosentan in Raynaud’s Syndrome and ulceration of the fingers (55). There is also evidence that cardiac fibrosis, occurred as a consequence of diabetes mellitus, is associated with accumulation of cardiac fibroblasts from endothelial cells. ET-1 has important role in this process (56).
Effects of endothelin on the cardiovascular system
ET-1 causes contraction of arteries and veins, which results in total peripheral resistance and mean arterial pressure increase. Contraction is often preceded by transient vasodilatation. These properties make ET-1 an important mediator in many cerebrovascular complications, including coronary artery diseases. Measured levels of ET-1, together with coronary magnetic resonance (CMR), have proved to be a useful prognostic parameter in microvascular obstruction (57). Increased levels of ET-1 can accelerate CMR microvascular obstruction and myocardial recovery assessment. High levels of this marker have proved to be associated with poor prognosis after myocardial infarction. Also, the concentrations of ET-1 can be used to identify patients who may yet develop microvascular obstruction. Therefore anti-endothelin therapy in ischemic heart disease treatment may offer additional benefits and effectively modify the progression of heart disease. The results of these studies indicate the value of measuring ET-1 concentration in patients with ischemic heart diseases (44,57). Future studies will show if increased concentrations of ET-1 lead to the development of microvascular obstruction, or it is just a secondary consequence of cardiac damage.
The role of endothelin in the treatment of hypertension
Many studies have shown that endothelin can be a pharmacological target for the hypertension treatment (58-65). ET receptor antagonists could be used as potential drugs for the hypertension treatment. However, ET receptor antagonists are in clinical use only for treatment of patients with pulmonary arterial hypertension (PAH). The reason for that are many possible side effects associated with ET receptor antagonists, as well as the existence of safer drugs for alleviating symptoms and for treating systemic hypertension. Adverse effects of ET receptor antagonists in clinical trials are common. The most frequently reported clinical adverse events are headache, peripheral edema, dizziness, nausea, nasal congestion, and dyspnea (58,59,62, 66). These appear to be a class effect, likely relate to vasodilation, and do not often lead to treatment withdrawal (66). Ambrisentan, an orally active, highly selective antagonist of the ETRA, is indicated for the treatment of pulmonary arterial hypertension (PAH). It has a low potential for drug-to-drug interactions and requires only once-daily administration. Ambrisentan was associated with a low risk of clinical worsening and of death (60). Bosentan, a dual endothelin receptor antagonist, is an effective and well-tolerated oral therapy for the management of pulmonary arterial hypertension (PAH; WHO group 1 pulmonary hypertension). Bosentan improves cardiopulmonary hemodynamics, exercise capacity, WHO functional class and quality of life, as well as delaying time to clinical worsening in patients with PAH (58,64). Recent studies have opened new possibilities for clinical use of ET receptor antagonists in the treatment of certain forms of drug-resistant hypertension (67,68).
The course of the disease and decrease of the symptoms were studied in a group of patients that showed resistance to the classic combination of three antihypertensive drugs (including diuretic), and underwent treatment with darusentan (selective ETRA antagonist) compared to the placebo group. Significant reduction of blood pressure was shown in the group that used darusentan when compared with the control group (67,68). Future studies should determine the effectiveness of darusentan for each patient with resistant hypertension (67).
The role of endothelin in the pathophysiology of atherosclerosis
It was shown that ET-1 is a key factor in the endothelial dysfunction development in various cardiovascular diseases including atherosclerosis. Endothelial dysfunction includes vasoconstriction, inflammation, thrombosis, blood vessel walls cell proliferation and fibrosis, thus leading to blood vessels clogging (microvascular obstruction). ET-1 is locally elevated in atherosclerotic plaques and circulating levels in the vascular tissue correlate with atherosclerotic lesions (69,70). Elevated levels of ET-1 induce gene expression for lipid metabolism products and thus lead to atherosclerosis development. Furthermore, increased expression of ET-1 aggravates atherosclerosis in Apo E-deficient mice (widely used hyperlipidemia model), and plaque formation and endothelial dysfunction can be restored in such atherosclerotic model using ETRA and even ETRB antagonists (71,72).
The role of endothelin in controlling utheroplacentary blood flow -occurrence of preeclampsia
Reduced uteroplacental perfusion is the initial event in pre-eclampsia, a disorder of pregnancy characterized by pregnancy-induced hypertension (≥ 140 mmHg systolic and/or ≥ 90 mmHg diastolic blood pressure), new on-set proteinuria (≥ 300 mg protein/day), and edema occurring in the second half of pregnancy (73). It is considered to be associated with increased synthesis of ET-1 which leads to vasoconstriction of uterine arteries and consequently to increased blood pressure. By monitoring the effect of antihypertensive drugs and ET-1 receptor antagonists it was proven that drugs that are currently used to treat pre-eclampsia, will not restore ET-1 induced contraction of the uterine arteries (74). Previous conclusion shows that they are not very effective. Only dihydropyridines partially relaxed previously contracted uterine artery. Thus, dihydropyridines could offer an interesting perspective for improving placental perfusion in pre-eclampsia (74).
Effects of endothelin on tumor development
ET-1 is the most potent cellular mitogen. Most of the patients with advanced bladder cancer develop lethal lung metastases. ET-1 has been implicated in this process, even though the mechanism(s) by which it promotes metastasis remains unclear. Evaluation of ET-1 mRNA and protein expression in four patient cohorts revealed that levels of ET-1 are higher in patients with muscle-invasive bladder cancers, which are associated with higher incidence of metastasis, and that high ET-1 levels are associated with decreased disease-specific survival (75). Consistent with its proinflammatory activity, tumor-derived ET-1 acts through ETRA to enhance migration of macrophages and induces expression of inflammatory cytokines. Tumor ET-1 expression and ETRA activity are necessary for metastatic lung colonization and this process is preceded by and dependent on macrophage infiltration of the lung. In contrast, tumor ET-1 expression and ETRA activity are less important in established primary or metastatic tumor growth. These findings strongly suggest that ETRA inhibitors might be more effective as adjuvant therapeutic agents than as initial treatment for advanced primary or metastatic disease (75).
Prostate cancer (PCA) is the most diagnosed cancer in men. The initial approach to advanced PCA is medical or surgical androgen deprivation therapy (ADT) or antiandrogens. Novel therapies complement the modest success that chemotherapy has demonstrated in recent years (76,77). These include agents targeting the epidermal growth factor receptor, endothelin receptor antagonists, angiogenesis inhibitors, immunomodulatory agents, immunotherapy, novel antiandrogens, and delivery of cytotoxic agents via therapeutic antibodies (76).
Endothelins are important regulators of angiogenesis and signal transduction. Generally, binding ET-1 to ETRA induces a survival pathway, whereas binding to ETRB promotes apoptosis. In PCA, hypermethylation of the ETRB promoter eliminates the negative growth response activating the ETRA pathway exclusively (76).
Atrasentan is an ETRA antagonist that prolonged time to PSA (prostate-specific antigen) progression when compared to placebo in a study conducted in asymptomatic men with PCA (75). ETRA induces a survival pathway, whereas binding to ETRB promotes apoptosis. Furthermore, combining atrasentan with docetaxel proved safe and effective with promising results (75,76).
Effect of endothelin on bones
A growing number of literature documents the presence and effects of endothelins in bones (77-81). Both ETRA and ETRB have been demonstrated in osteoblastic cells by ligand binding. Major signal transduction pathways for endothelin in bone cells appear to be stimulation of phospholipid turnover, activation of A, C and D phospholipases, stimulation of calcium flux from intracellular and extracellular stores and activation of tyrosine kinases (78). Endothelins also modulate calcium signaling elicited by other agents in osteoblastic cells. The parathyroid hormone-stimulated calcium transient in UMR-106 cells is enhanced by endothelins, acting through an ETBR, whereas the parathyroid hormone-stimulated increase in cAMP is inhibited by endothelins (78). Phenotypic responses to endothelin-1 include changes in alkaline phosphatase activity, stimulation of osteocalcin and osteopontin message, stimulation of collagen and noncollagenous protein synthesis, inhibition of osteoclast motility and stimulation of prostaglandin-dependent resorption (78). It is proven that ET-1 inhibits alkaline phosphatase in mouse osteoblastic cells and has biphasic effects on alkaline phosphatase in human osteoblastic cells (78). Similar to its effects in other tissues, ET-1 stimulates calcium/phospholipid signaling pathways in bones. Treatment with ET-1 increases calcium and inositol trisphosphate in the endothelial cells in human parathyroid glands. Endothelin was found to inhibit the secretion of parathyroid hormone from human adenoma cells and in an in vivo parathyroid gland perfusion model (78). These findings suggest that endothelin could have a regulatory function in parathyroid hormone secretion. Endothelin-1 also enhances the interleukin-1-induced increase in interleukin-6 (78-81). Inflammatory markers are key players in bone biology and are involved in the regulation of osteocytes; as a result, the dynamic balance of bone formation and resorption are influenced by inflammatory markers (82). It has been shown that local administration of endothelin receptor antagonists reduces edema, neutrophil infiltration and production of inflammatory mediators (83). Endothelins can also potentially affect calcium metabolism by inhibiting the secretion of parathyroid hormone (78). These observations raise the need for further studies to elucidate the physiologic role of endothelins as possible endocrine, paracrine or autocrine factors in calcium metabolism and even the possible therapeutic applications of receptor-selective endothelins and antagonists in disorders of bone and calcium metabolism.
Conclusion
Within 20 years after its initial discovery it has become clear that endothelin is not merely a vasoconstrictor but that it is a multifunctional peptide with cytokine-like activity which affects almost all aspects of cell function (84).
Many studies point towards a beneficial effect of the blockade of ET receptors on vascular function in these diseases. However, despite of this enormous experimental work, the clinical use of ET receptor antagonists is limited to pulmonary arterial hypertension and digital ulceration in SSc to date. The future is not as pessimistic as a new era is being unveiled with the generation of powerful genetic models, which will allow the dissection of the complex physiology of the ET-1 system, and also with the recent discovery of specific roles of ET-1. In order to translate this new knowledge to the clinical practice, more experimental studies together with the commitment of the pharmaceutical industry to develop the corresponding preclinical and clinical trials are required.