Introduction
Hereby we report two hemoglobin anomalies not previously described in subjects of Central European origin, namely hemoglobin Hasharon, an alpha chain variant, in two non-related patients,and hemoglobin NYU, a delta chain variant, in one patient. The patients were treated at the University Clinics Bonn for illnesses unrelated to these hemoglobin anomalies. The attending physicians had requested HbA1c analysis, which we performed in our laboratory using high performance liquid chromatography (HPLC) (Variant, Biorad).
Hb Hasharon [a(2)47(CD5)Asp®Hisb2] is a hemoglobin anomaly with a mutation in the alpha globin. It was first described in 1968 as Hb Sinai in a family of Jewish origin (1). In the literature, sporadic incidences of Hb Hasharon have been reported from Israel, Italy, Greece and in Latin Americans of Mediterranean origin (2-4). We detected heterozygous Hb Hasharon in a 60-year old female patient and a 74-year old male patient. Both patients have German ancestors.
Hb NYU [d12(A9)Asn®Lys] is a hemoglobin anomaly with a mutation in the delta globin. It was first described in 1969 in two families of Russian-Jewish origin (5). In the literature, this mutation has to date been reported in Jewish patients and patients of Italian and Greece origin (6,7). We detected Hb NYU in a 72-year old female patient of German origin.
Materials and methods
The study was performed at the University hospital Bonn from July 2010 until October 2010. Through informed consent we obtained EDTA-blood and serum from all patients. Hemolysates from EDTA-blood were analyzed for high performance liquid chromatography analysis, capillary zone electrophoresis and hemoglobin electrophoresis. EDTA-blood was analyzed for gene sequencing on a genetic analyzer and for blood cell counts. Serum samples were analyzed for serum iron, ferritin, soluble transferrin receptor, haptoglobin, lactate dehydrogenase, aspartate amino transferase and potassium on a clinical chemistry analyzer.
HPLC was performed with the Beta-Thal program of Variant II (Biorad, Munich, Germany). Hemoglobin electrophoresis in the acidic and the alkaline range was carried out with the Hydrasys system (Sebia, Fulda, Germany) and hemoglobin capillary zone electrophoresis with the Capillarys system (Sebia, Fulda Germany). Complete blood count was analyzed with the XE-5000 analyzer (Sysmex, Norderstedt, Germany), clinical chemical analysis was carried out with Dimension Vista 1500 (Siemens, Eschborn, Germany).
DNA was isolated from peripheral blood leukocytes. Polymerase chain reaction (PCR) and gene sequencing of alpha, beta and delta globins was performed by standard procedures. In brief, human α1- and α2-globin genes (HBA1, HBA2) were selectively amplified with a universal forward primer (5’-CCAAGCATAAACCCTGGCGC-3’) and a reverse primer complementary to a nonhomologous part of their 3’ untranslated region (α1: 5’-CACGGGGGTACGGGTGCAG-3’; α2: 5’-AGGAAGGGCCGGTGCAAGG-3’). The HBB gene was amplified with primers 5’-GTCAGGGCAGAGCCATCTATT-3’ and 5’-GCACTGACCTCCCACATTCC-3’ and, in case of the HBD gene, primers 5’-CACTGGAGCAGGGAGGACAG-3’ and 5’-AAGCCATACCCTTGAAGTAGGC-3’ were used. PCR primers also served as sequencing primers using a 3130/x genetic analyzer (Applied Biosystems, Darmstadt, Germany).
Results
Hb Hasharon
In the female patient, the complete blood count was normal with RBC 5.0 x 1012/L (4.1-5.1 x 1012/L), Hb 14.3 g/dL (12.3-15.3 g/dL), hematocrit 0.41 L/L (0.35-0.47 L/L), MCV 83 fL (80-96 fL), MCH 29 pg (27 – 34 pg); clinical chemical analysis revealed normal haptoglobin values, there was no indication of hemolysis and ferritin value was within the reference range. HPLC showed an 18.2% peak at 4.17 minutes retention time. Capillary zone electrophoresis showed a peak of 20.5% in zone 4 and a small peak of 0.5% in zone 1. Hemoglobin electrophoresis revealed in the acidic range a band directly after HbA0 to the anode. In the alkaline range, a band was apparent directly after in direction to the anode (Figure 1). Gene sequencing resulted in [a(2)47(CD5)Asp®Hisb2]. In the male patient, the complete blood count was also normal with RBC 3.9 x 1012/L (4-5.65 x 1012/L), Hb 13.3 g/dL (12.5-17.2 g/dL), hematocrit 39% (37-49%), MCV 99 fL (80-101 fL), MCH 34 pg (27-34 pg). In both patients, concomitant alpha-thalassemia could be excluded.
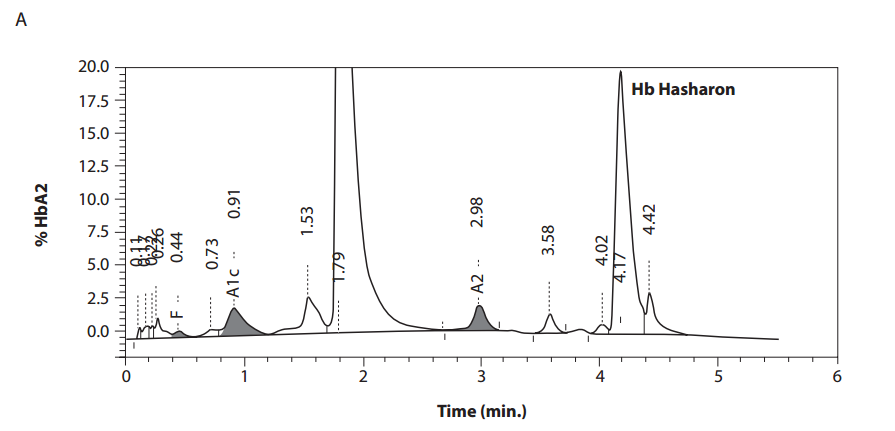
Figure 1. Detection of Hb Hasharon by HPLC (A).
B 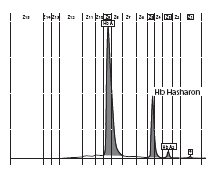
C 
D 
Figure 1. Capillary zone electrophoresis (B), acidic Hb electrophoresis (C) and alkaline Hb electrophoresis (D).
Hb NYU
The patient’s complete blood count was normal with RBC 4.6 x 1012/L (3.85-5.2 x 1012), Hb 13.4 g/dL (11.8-15.8 g/dL), hematocrit 0.41 L/L (0.355-0.455 L/L), MCV 89 fL (80-101 fL), MCH 29 pg (27-34 pg). Clinical chemical analysis revealed normal haptoglobin levels and there was no indication of hemolysis, while ferritin value was normal. In HPLC, a 1.1% peak was noted at 3.65 minutes retention time. In capillary zone electrophoresis, a peak of 0.9% was noted between zone 1 and zone 2 as well as a low HbA2 of 1.2%. In hemoglobin electrophoresis, a band was noted in the acidic range directly after HbA0 to the cathode, while in the alkaline range, a band was noted directly after the HbA0 band to the anode (Figure 2). Gene sequencing resulted in [d12(A9)ASN®Lys].
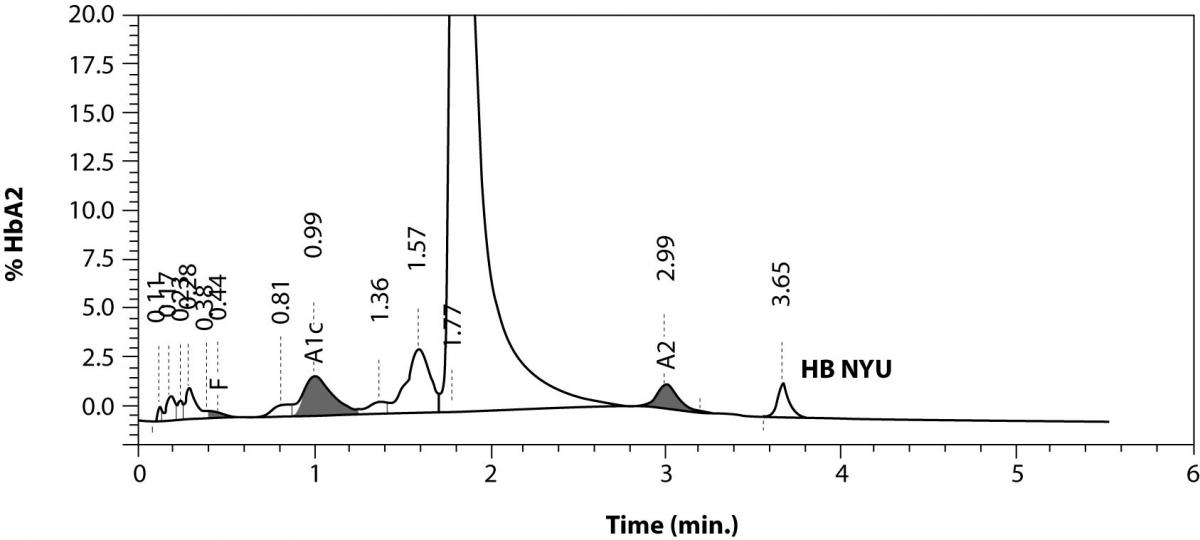
Figure 2. Detection of Hb NYU by HPLC (A).
B 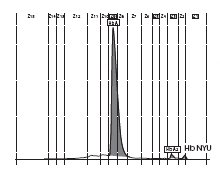
C 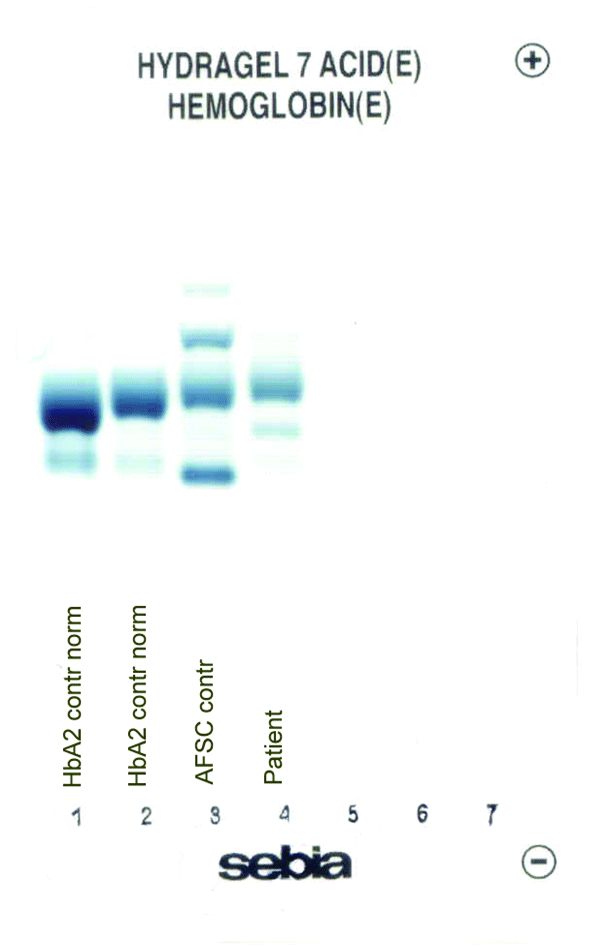
D 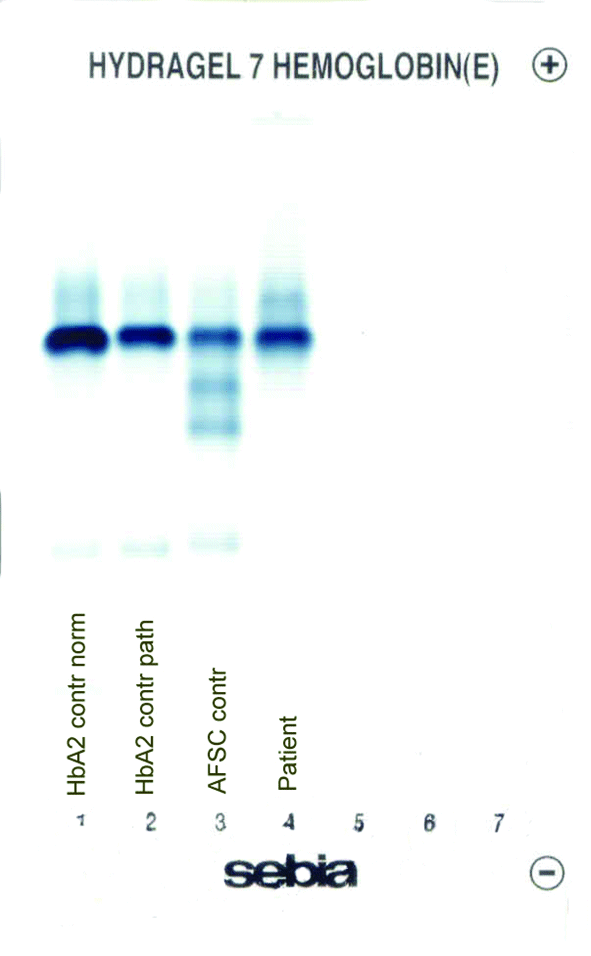
Figure 2. Capillary zone electrophoresis (B), acidic Hb electrophoresis (C) and alkaline Hb electrophoresis (D).
Discussion
Hb Hasharon is a hemoglobin variant with an alpha globin mutation with no clinical relevance (8). However, with concomitant alpha-thalassemia it can lead to hemolytic anemia (3). In both of our patients, clinical chemical laboratory parameters as well as complete blood count were normal. Also, no hemolysis was found. Hb NYU is a rare hemoglobin anomaly with the delta globin gene affected. In healthy adults, delta globin is only present in the HbA2 fraction, which normally constitutes up to a maximum of 3.5% of total hemoglobin. Clinically relevant cases of anemia or hemolysis have only been described with concomitant thalassemia (9). In our female patient, all clinical chemical parameters as well as the complete blood count were normal. Hemoglobin Hasharon and hemoglobin NYU have to date only been reported in patients of Jewish and/or Mediterranean origin. Following intensive research, our patients confirmed their German origin with no ancestors of Jewish or Mediterranean origin. No hemoglobin anomaly had been reported in any of the families. Unfortunately, due to the advanced age of our patients we were unable to analyze blood from immediate ancestors.
Neither Hb Hasharon nor Hb NYU have been reported in subjects of German origin to date.