Introduction
The proces of lipid peroxidation is one of oxidative conversion of polyunsaturated fatty acid to cytotoxic products, and one of them is the malondialdehyde (MDA) (1). Possible causes of oxidative stress in diabetes are as follows: a) excessive production of reactive oxygen species (ROS), particularly superoxide anions (O2-); b) reduced expression/activation of endothelial NO synthase; c) impaired expression/ activation of superoxide dismutase (SOD); d) reduced activity of antioxidative enzymes catalase (CAT) and glutathione peroxidase (GPx); e) reduced level of antioxidants: glutathione, a-tocopherol and ascorbate; f) increased nonenzymatic protein glycosylation and formation of the so-called advanced glycosylation end products (AGE); g) enhanced glucose autooxidation; h) hyperactivity of polyol pathway and sorbitol production. Reactive oxygen species (ROS) may alter the function of vascular endothelium indirectly by increased production of AGE or by increased oxidation of low density lipoproteins (LDL). The most important ROS compound is superoxide anion (O2-) as it causes vascular smooth muscle contraction (2). Major consequences of oxidative stress are destruction of biomembranes, nucleic acid damage, enzyme inhibition, protein degradation and lipid peroxidation. Chemical modification of amino acids in proteins during lipid peroxidation results in the formation of lipooxidative products that serve as markers of oxidative stress in vivo. Malondialdehyde (MDA) and 4-hydroxynonenal (HNE) are well characterized oxidation products of polyunsaturated fatty acids (3). MDA reacts with vascular proteins, e.g. collagen, and leads to changes in their structure (4). Many studies showed considerably elevated MDA concentration in diabetes mellitus (5). Faure et al. claim that glycemia controlis necessary to reduce lipid peroxidation, i.e. MDA levels (6). Acarbose is the first drug of choice in the treatment of diabetes mellitus Type 2 (7). Therefore, the aim of this study was to evaluate oxidative stress in alloxan-induced non-obese diabetic (NOD) mice by measuring MDA levels in their liver homogenate, and investigate the effect of acarbose on the concentration of this parameter of lipid peroxidation.
Material and methods
Experimental animals
The mice selected for this study were of NOD (non-obese diabetic) strain, age 3-4 months and 23-30 g body weight, bred at the Laboratory for Molecular Endocrinology and Transplantation, Ruđer Bošković Institute, Zagreb, Croatia. The animals were kept in metabolic cages at the temperature of 22-24 oC and 12 h light/dark cycle, and were fed standard laboratory chow (Pliva Company, Zagreb, Croatia) with water available ad libitum. In our experiment, diabetes was induced in mice by i.v. administration of alloxan-monohydrate (Sigma, St. Louis, USA) in a dose of 75 mg/kg b.w. seven days before the onset of acarbose treatment. The animals were divided into four groups, with 6 mice in each: Control, healthy group of non-obese mice (C), control, healthy group of non-obese mice on 7-day acarbose treatment in a dose of 25 mg/100 g chow (C/A), group of non obese diabetic (NOD) mice (D), a group of NOD mice treated for 7 days with acarbose dose of 25 mg/100 g chow (D/A).
Immediately before sacrificing during ether narcosis, venous blood was collected from mice for determination of glucose concentration.
Preparation of liver homogenate
Following sacrificing under ether narcosis, liver was extracted from mice, multiply rinsed in physiologic solution, and kept cool until analysis. The liver was weighed, and then homogenized by Ultra-Turax homogenizer, type TP 1812 NR 3219 (three times at 13,000 rpm/30 sec). 100 g/l homogenate concentration was prepared using 0.14 M of KCl solution and continuous cooling in ice water. Liver homogenate was then centrifuged at 12,000 g for 30 minutes at +4 oC in Hettich EBA 12 R Eppendorf centrifuge. After centrifugation, it was stored at -20 oC until analysis. The liver homogenate obtained in this manner was used for determination of MDA concentration.
Determination of MDA concentration
Lipid peroxidation was established by measuring malondialdehyde (MDA) in mouse liver homogenates using thiobarbituric acid (8). The method is based on the following principle: the heating of thiobarbituric acid and polyunsaturated fatty acids in acid media results in the formation of pink-colored secondary product of MDA whose absorption is measured spectrophotometrically at 532 nm wavelength. The absolute amount of MDA was read from a calibration diagram prepared from different dilutions of the primary standard of 1,1,3,3-tetrametoxipropane (TMP).
Glucose level determination
Blood glucose was measured in mice by the glucose oxidase-peroxidase (GOD-PAP) (9) method using the Trace company reagents. Absorbance of the sample was read at 500 nm wavelength against the blank and the standard.
Statistical method
Mean value and standard deviation (SD) were calculated using a computer and MicrosoftÒ Excel application. The level of significance (p) was calculated using Student t-test and the same application. A change was considered statistically significant if p<0.05.
Results
Serum glucose level in mice
Table 1 shows glucose concentration in mouse sera determined on day 7 since the beginning of acarbose treatment. Glucose concentration in the control, healthy group of mice (group C) was 4.63 ± 0.30 mmol/l. In the control group with acarbose dose of 25 mg/100 g standard laboratory chow (group C/A), glucose concentration was 2.85 ± 0.42 mmol/l, i.e. glucose produced a significant hypoglycemic effect (p<0.001). NOD mice (group D) had significantly higher glucose level compared to the control, healthy group (group C) (p<0.01). The serum glucose level in group D was 31.28 ± 4.54 mmol/l, while in acarbose treated NOD mice given the dose of 25 mg/100 g standard laboratory chow (group D/A) glucose concentration was 16.44 ± 9.5 mmol/l, which was also significantly lower compared to the diabetic group D (p<0.05).
MDA concentration in liver homogenate of NOD mice
MDA concentration determined in liver homogenates of NOD mice and results are also presented in Table 1.
Table 1. Glucose concentration in the serum (mmol/L) and MDA concentration in the liver of NOD mice (mM/g liver tissue)
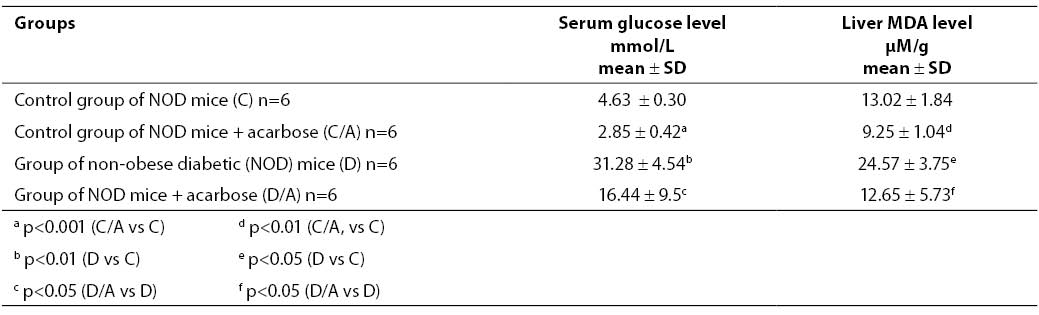
The MDA concentration measured in the liver of control, healthy NO mice (group C) was 13.02 ± 1.84 mM/g. A significantly lower MDA concentration, i.e. 9.25 ± 1.04 mM/g (p<0.01) was measured in the group of healthy NO mice subjected to 7-day acarbose treatment in the dose of 25 mg/100 g standard laboratory chow (group C/A). The group of non-obese diabetic (NOD) mice (group D) had a significantly higher MDA concentration compared to the control, healthy group (p<0.05). The change in MDA concentration measured in the group of NOD mice subjected to acarbose treatment in the dose of 25 mg/100 g standard laboratory chow (group D/A) was significantly lower compared to group D (p<0.05) (12.65 ± 5.73 mM/g vs. 24.57 ± 3.75 mM/g).
Discussion
Oxidative stress is characterized by increased production of free radicals and/or reduced activity of antioxidative defense. In an experimental model of diabetes, oxidative stress was induced by glucose autooxidation and protein glycation (10). Considerable attention is currently dedicated to oxidative stress as a process that is probably responsible for the occurrence of complications in diabetes (atherosclerosis, nephropathy, neuropathy and retinopathy) (11,12). One of the consequences of oxidative stress is also lipid peroxidation. Determination of lipid peroxidation parameters includes measurement of the concentration of lipid peroxides, conjugated diens and malondialdehyde (MDA); the last of these three parameters is a highly toxic final product of lipid peroxidation, probably the result of non-enzymic oxidative degradation of lipids. Altomare et al. (13) compared MDA concentrations in groups of diabetic patients with poorly and those with well controlled glycemia, and compared their results with healthy, normoglycemic individuals. Their results showed substantially elevated plasma lipid peroxide levels in patients with poorly controlled glycemia in comparison to the two other subject groups. An experiment conducted by Armstrong et al. (14) showed that strictly controlled diet in diabetic patients is sufficient to decrease MDA concentrations.
Hyperglycemia in diabetes is a primary cause of oxidative stress and lipid peroxidation. Actually, it brings about changes in cellular redox status via several various mechanisms: it changes the regulation of protein tyrosine kinase and of protein kinase C, it leads to sorbitol accumulation and increases NADH/NAD+ ratio and decreases NADPH/NADP+ ratio via hyperactivity of the sorbitol (polyol) pathway. The overall ensuing effect is cytosolic redox imbalance, the condition called pseudohypoxia, due to elevated NADH/NAD+ ratio that conceals tissue hypoxia (15). Administration of hyperglycemia reducing medications (particularly post-prandial) is important to lessen diabetic complications caused by oxidative stress. Therefore we investigated in this study the hypoglycemic effect of acarbose, as well as its effect on oxidative stress, i.e. lipid peroxidation in NOD mice. Acarbose is a pseudooligosaccharide obtained as a secondary metabolite from Actinomycetales culture. An acarbose molecule consists of an unsaturated cyclohexitol residue bound to amino saccharide and two glucose residues via a-1,4 glycoside bonds. Its antihyperglycemic effect is based on the fact that acarbose is a competitive inhibitor of intestinal a-glucosidases that comprise glucoamylases, saccharases, maltases and a-dextrinases, the enzymes that are necessary for degradation of starch, saccharose and maltose (16). Acarbose binds to the above a-glucosidase with the affinity which is 10,000 to 100,000-fold higher than that of, e.g., saccharose. Inhibition of this enzymic activity by acarbose reduces the rate of monosaccharide production and their absorption in the intestine. A new investigation confirm beneficial effect of acarbose on postprandial hyperglicaemia in prediabetic and Type 2 diabetic patients (17). Acarbose, also, might reduce macrovascular complication by avoiding endothelial injury in postprandial hyperglycemic status (18). There is growing evidence that oxidative stress and inflammation are involved in the pathogenesis of cardiovascular disease in diabetes mellitus. However, it is still unclear whether this phenomenon can be controlled in clinical practice by modulating postprandial hyperglycemia. In study of Assaloni et. al. shows that controlling postprandial hyperglycaemia with mitiglinide significantly improves the cluster of oxidative stress and inflammation markers that are increased in the postprandial state in diabetic patients (19).
A significantly elevated serum glucose level (p<0.01) and MDA concentration in the liver (p<0.05) is reported in our study in NOD mice with alloxane induced diabetes (D), compared to the control, healthy group (C), the fact which confirmed oxidative stress in the group of non-obese diabetic mice. Hypoglycemic effect of a low acarbose dose (25 mg/100 g standard laboratory chow) (p<0.05) was experimentally proven; also, a favorable effect of acarbose on oxidative stress was recorded since MDA concentration in NOD mice after 7-day acarbose treatment (group D/A) was significantly lower (p<0.05) compared to the group of non-obese diabetic mice (group D). The favorable effect of acarbose on MDA concentration may be accounted for by its antihyperglycemic action.