Introduction
The incidence of venous thromboembolism (VTE) is considerably lower in children than in adults. It is estimated that the incidence of VTE in children is one thenth of that in adults (1). Investigations of VTE in childhood have shown that most children with deep venous thrombosis (DVT) and pulmonary embolism (PE) have underlying hereditary thrombophilia (2-5). Additional risk factors (infection, immo bilization, trauma, surgery, malignancy) in combination with genetic factors of thrombophilia significantly increase the risk of VTE occurrence. Each case of VTE in childhood requires thorough clinical approach and complete laboratory diagnosis that will suggest whether it represents an isolated event, genetic thrombophilia or a combination of genetic and acquired risk factors for the occurrence of this disease.
Materials and methods
All coagulation tests were performed on a Behring Coagulation System analyzer (BCS, Dade Behring, GmbH, Austria) with commercially available reagents. Activated protein C resistance (APCR) was determined by the coagulation-based method using ProC® Global reagent, and lupus anticoagulant (LAC) by use of LA1/LA2 reagents from the same company. Enzyme immunoassay was used to analyze anticardiolipin antibodies (ACA) of IgG and IgM classes (ELISA, Orgentec Diagnostika, GmbH, Germany) and antibodies to double-stranded DNA (anti-dsDNA) (ELISA, Pharmacia Diagnostics, Sweden). Detection of mutations for factor II (G20210A) and factor V (G1691A, Factor V Leiden) was performed by melting curve analysis using commercially available kits (PCR-LightCycler Mutation Detection Kits, Roche, Germany) on a Roche LightCycler® apparatus. The polymorphisms C667T in the gene for methylenetetrahydrofolate reductase (MTHFR) and 4G/5G in the gene for plasminogen activator inhibitor-1 (PAI-1) were determined using PCR-RFLP method.
Case report
A 14-year-old boy presented to emergency pediatric clinic of the University Department of Pediatrics for the right lower extremity pain and swelling, where he underwent medical examination and was admitted to the Department. For a month before, the boy was on outpatient antibiotic treatment for infection. Two days before hospitalization, in addition to pain in the right leg, rigidness and swelling of the right leg occurred. Because of suspect DVT, duplex doppler of deep veins was obtained on day 1 of hospitalization to prove DVT of the leg veins and popliteal vein. Therapy with low-molecular-weight heparin (Fragmin®, 5000 IU, s.c.) was introduced. On day 5, the patient felt dyspnea and pain in the left thorax. The diagnosis of PE complicated with pulmonary infarction was made by magnetic spiral computerized tomography of the thorax and that night the patient was transferred to coronary unit of the University Department of Medicine. Therapy with unfractionated heparin was introduced (at an initial dose of 25,000 IU/24 h, i.v.). Gradual good clinical response was achieved with target values of activated partial thromboplastin time (APTT) from 1.9 to 2.7. After two days, an oral anticoagulant (Marivarin®) was introduced in therapy, along with unfractionated heparin. APTT and prothrombin time (PT) were determined daily according to the protocol for monitoring of double anticoagulant therapy for VTE. After eight days of hospital stay, the treatment was continued at University Department of Pediatrics with unfractionated heparin and Marivarin®. Unfractionated heparin was excluded after eleven days and low-molecular-weight heparin (Fragmin®, 5000 IU, s.c.) was introduced again with Marivarin®. This therapy was continued for one month. Upon discharge from the hospital, the treatment has been continued with Marivarin® alone until now, with regular outpatient control of PT (portion, INR) at hematology clinic of the University Department of Pediatrics.
The patient's personal and family history revealed him to be the first-born child from the first pregnancy. After this pregnancy, the patient's mother had a spontaneous abortion. The boy had normal perinatal and growth history. In his family history, his paternal grandmother suffered cerebrovascular insult, and his mother's parents were cardiovascular patients. The patient's mother suffered from diabetes mellitus. There was no family history of DVT in young relatives.
Results
Results of all laboratory analyses according to time points are presented in Table 1.
Table 1. Results of laboratory analyses performed upon admission to emergency pediatric clinic (A), on hospitalization day 1, immediately after the diagnosis of DVT had been established and therapy with low-molecular-weight heparin initiated (B), on hospitalization day 5 when PE developed and therapy with unfractionated heparin was initiated (C), and nine weeks of admission to emergency pediatric clinic, on Marivarin® therapy (heparin therapy excluded) (D)
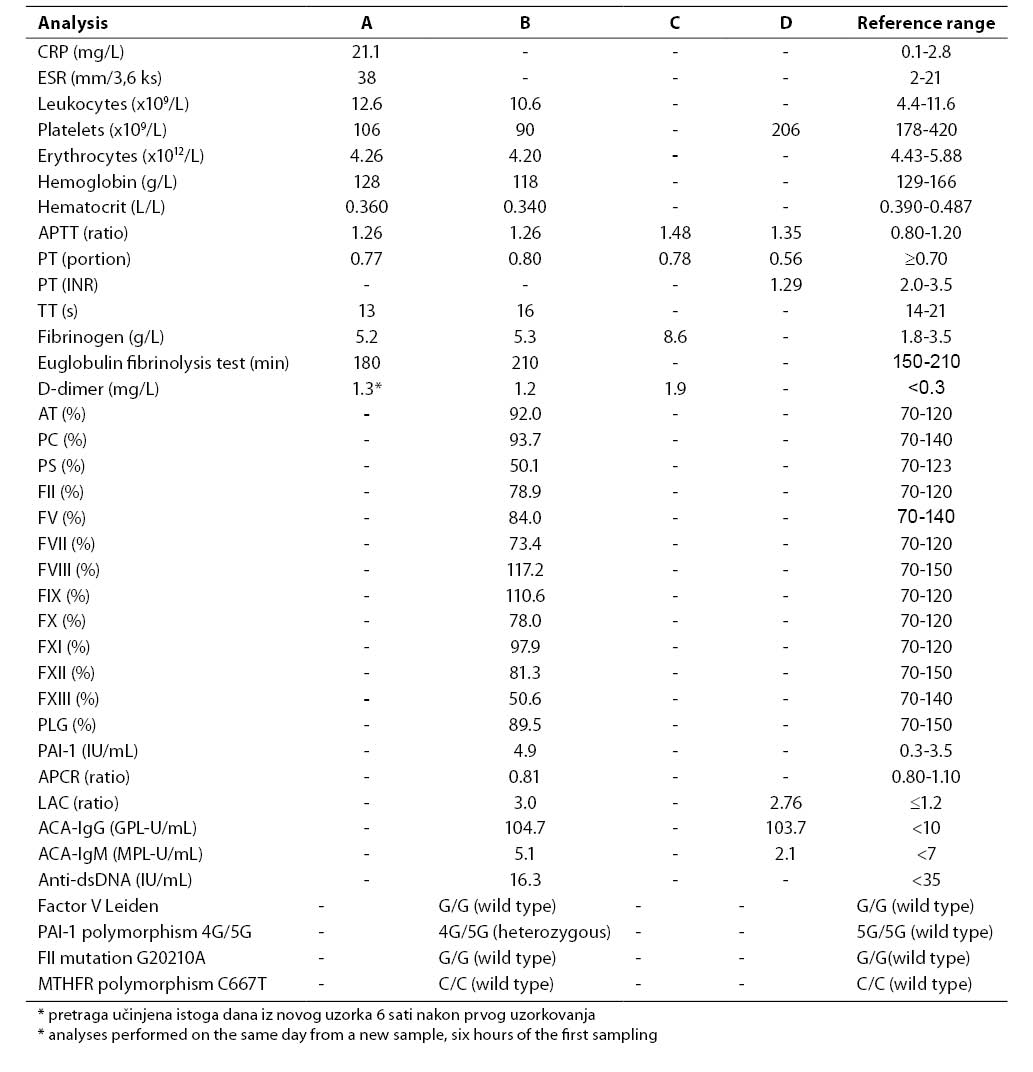
Common hematology and coagulation analyses performed upon the patient's admission to emergency pediatric clinic showed a slightly prolonged APTT (1.26), elevated fibrinogen concentration (5.2 g/L) and decreased platelet count (106x109/L) (Table 1, column A). Because of suspect DVT, the concentration of D-dimer was determined on the same day and was found to be increased (1.3 mg/L). Results of extended laboratory analyses that included studies of trombophilic factors, performed on hospitalization day 1, immediately after the diagnosis of DVT had been established, are presented in Table 1, column B. Platelet count remained low (90x109/L). The activities of coagulation factors FII to FXII were within the reference ranges, and the activity of FXIII was decreased (50.6%). Determination of the activities of the coagulation inhibitors antithrombin (AT), protein C (PC) and protein S (PS) showed a decreased activity of PS (50.1%), while the activities of AT and PC were within the reference ranges. The ProCGlobal coagulation-based assay for APCR yielded a borderline value (APCR ratio 0.81). Molecular diagnosis excluded G1691A mutation for coagulation factor V (factor V Leiden) as the most common possible cause of APCR. Molecular diagnosis of other hereditary thrombophilic factors excluded G20210A mutation in the gene for coagulation factor II (prothrombin) and C667T polymorphism in the gene for MTHFR, while demonstrating polymorphism for PAI-1 (genotype 4G/5G). Plasma concentration of PAI-1 was increased (4.9 IU/mL).
Antiphospholipid antibodies, LAC and significantly elevated titer of ACA-IgG, were demonstrated in the patient. Since the analyses of antiphospholipid antibodies (LAC, ACA IgG and IgM) were performed when therapy with low-molecular-weight heparin had already been introduced, the same tests were repeated after nine weeks when heparin was excluded from therapy protocol, and the patient was on therapy with Marivarin® still remained elevated (Table 1, column D).
After the diagnosis of PE was established on day 5 of his hospital stay, the concentrations of D-dimer and fibrinogen (8.6 g/L) showed further increase (Table 1, column C).
Discussion
Presenting this patient we report on the sequence of clinical and laboratory diagnosis from the patient’s presenting to the emergency pediatric clinic, admission to the ward, establishment of the diagnosis, and identification of the possible cause of VTE.
It is known that laboratory diagnosis of thrombophilia is not recommended to perform in the acute stage of disease or during anticoagulant therapy (5,6). In this case, it was not possible since the patient was maintained on anticoagulant therapy from the first day of hospitalization until now, and because of the established trombophilic factors he is a candidate for long-term therapy. Laboratory analyses that included investigation of hereditary and acquired thrombophilic factors were performed on hospitalization day 1, immediately after the diagnosis of DVT had been made and therapy with low-molecular-weight heparin initiated. Determination of antiphospholipid antibodies that suggested the presence of antiphospholipid syndrome (APLS) in the patient and the results of which can be influenced by heparin therapy, was repeated after therapy with unfractionated and low-molecular-weight heparin had been excluded.
In the acute stage of thrombosis, the patient had a decreased platelet count. Transiently decreased platelet count in patients with DVT is often the result of the increased platelet consumption during the acute stage of thrombosis (6). During treatment, platelet count reached values within the reference range (175-234x109/L, data not presented). The decreased activity of factor XIII in the acute stage of disease is the result of consumption in the process of coagulation activation (6). On initial clinical suspicion of DVT, the concentration of D-dimer was elevated (1.3 mg/L), with further increase when PE developed (1.9 mg/L). These results suggest clinical significance of D-dimer determination in the diagnosis of DVT and PE (6,7).
Antiphospholipid antibodies, LAC and ACA IgG, were demonstrated on two independent measurements nine weeks apart. These results, along with clinical presentation of VTE, suggest the presence of APLS in this patient. The most common clinical manifestation of APLS is DVT of lower extremities, complicated with PE in one half of patients (8). In subjects with APLS, thrombosis may occur spontaneously or in the presence of other risk factors, genetic or acquired. The diagnosis of APLS is established if at least one clinical criterion (venous or arterial thrombosis, spontaneous abortion) and one laboratory criterion (increased ACA IgG and/or IgM, presence of LAC on two independent measurements at least six weeks apart) are met (9). APLS is the most common cause of acquired thrombophilia and the syndrome association with frequent occurrence of VTE has been demonstrated in a number of studies (10-16).
Molecular diagnosis of hereditary thrombophilic factors confirmed PAI-1 polymorphism (genotype 4G/5G), with regular reports for other molecular markers. Although PAI-1 polymorphism is not considered as an independent risk factor for VTE, some literature data suggest an additional role of this polymorphism in VTE occurrence in subjects who are carriers of genetic or acquired thrombophilic factors (17-20). PAI-1 is a physiological regulator of the fibrinolytic system, whose binding to tissue plasminogen activator inhibits activation of plasminogen into plasmin and thus inhibits fibrinolytic activity. In subjects with 4G/5G polymorphism in PAI-1 gene, elevated plasma concentration of PAI-1 acts as a prothrombotic factor because of disorder in the fibrinolityc system (6,20,21). The increased plasma concentration of PAI-1 (4,9 IU/mL) is in accordance with established 4G/5G genotype for PAI-1 and suggests that PAI-1 polymorphism, besides APLS, may have played an additional role in VTE occurrence in our patient.
Coagulation-based assay for APCR suggested the borderline resistance to APC (APCR ratio 0.81). Molecular diagnosis excluded G1691A mutation for coagulation factor V (factor V Leiden), that is in 90% of cases the cause of resistance to APC. Because it is the case of a patient with the antiphospholipid antibodies LAC and ACA, the possible explanation for decreased APCR is an interference of antiphospholipid antibodies in the activated protein C system (5,8).
Determination of activities for the coagulation inhibitors antithrombin (AT), protein C (PC) and protein S (PS) showed a decreased activity of PS (50.1%), with the activities of AT and PC within the reference range. However, interpretation of PS values during acute disease is difficult. About 60% of PS in the circulation is bound to C4b-binding protein (C4b-BP), while 40% of PS is found in the free form and acts as a cofactor of APC in the inactivation of factors Va and VIIIa. C4b-BP is an acute phase reactant, whose plasma concentration significantly increases during the acute stage of disease, thus the portion of bound PS increases and the portion of free PS decreases. Therefore, a decreased activity of PS measured in our patient could have been the result of elevated C4b-BP concentration in the infection that had been present several weeks before DVT as well as the result of elevated C4b-BP concentration in the acute stage of thrombosis (5,6,22). On the basis PS activity measurement in the acute stage of thrombosis it is not possible to establish whether there is a hereditary deficiency of PS or acquired deficiency caused by infection and thrombosis. The diagnosis of hereditary deficiency of PS requires determination of the activity and concentration of free PS in plasma upon completion of treatment for DVT.
In conclusion, laboratory diagnosis of thrombophilia in the described patient confirmed APLS as a risk factor for VTE. Additionally, although PAI-1 polymorphism is not considered as an independent thrombophilic factor, it is possible that in patients with established thrombophilic factors such as APLS this polymorphism may play an additional role in VTE occurrence. Infection and prolonged bed rest that preceded thrombosis could have been additional risk factors for VTE occurrence in this patient. The reported case supports the current concept according to which VTE most commonly results from interaction of genetic and acquired risk factors.