Introduction
Glucose, apart from being energy source via glycolysis and citrate cycle, also participates in many other intracellular metabolic pathways such as pentose-phosphate shunt, glycogenesis, nucleotide sugar synthesis, etc. One of them is the hexosamine biosynthesis pathway (HBP) and its end-product: protein-associated O-glycosylation (O-GlcNAc). In the last few years, hexosamine pathway and O-GlcNAc became an intensively investigated topic, in particular because they might interfere with kinases and phosphorylation in a number of signaling pathways (1). O-glycosylation takes place and remains inside the cytoplasm and the nucleus on susceptible target proteins due to a specific and reversible enzymatic transfer of the hexosamine pathway metabolite: UDP-N-acetylglucosamine to the OH group of serine or threonine amino acids of these proteins.
O-GlcNAc was first described in 1984 (2) and since then its importance in several cellular processes has been recognized, such as nutrient sensing (3), cell-cycle regulation (4), or modification of several transcriptional factors (5-9). Given that O-GlcNAc has such widespread functionality it is not surprising that it has been associated with the development of numerous patho-physiological processes. The most obvious and well known significance of O-GlcNAc has been established in chronic complications of diabetes mellitus and insulin resistance (reviewed in 10); however, stress adaptation, especially regarding the heart, has lately also come in the spotlight (11,12). It is somewhat paradoxical that while, on the one hand, the effect of increased flux through the hexosamine pathway in diabetes is deleterious, on the other hand increased O-GlcNAc seems to be beneficial in joint diseases such as osteoarthritis, or in acute stress-situations such as an ischemic heart attack (13,14). Our goal was to give an extensive summary on the mechanisms and involvements in signaling pathways, to review the current understanding of the role of GlcNAc in certain diseases and to get a deeper insight of the Janus faced properties of O-GlcNAc.
HBP pathway
Approximately 2-4% of the glucose entering the cell is metabolized through the hexosamine biosynthetic pathway (HBP). The key enzyme is glutamine-fructose-6P amidotransferase (GFAT) which catalyzes the L-glutamine + D-fructose-6P = L-glutamate + D-glucosamine-6P reaction. This reaction can be bypassed by directly adding glucosamine to cells and thus increasing the HBP flux. Following the two subsequent downstream metabolites (N-acetylglucosamine-6P, N-acetylglucosamine-1P), the end-product of HBP is UDP-N-acetylglucosamine (UDP-GlcNAc).
UDP-GlcNAc is utilized in peptidoglycan synthesis, in lipo- and mucopolysaccharide synthesis, N-type (linked to the amide “N” on the sidechain of Asn) or O-type proteoglycan biosynthesis (most commonly, first N-acetylgalactosamine (galNAc) is linked by an α-glycosidic linkage to the OH group of Ser/Thr, then additional carbohydrate molecules are attached such as GlcNAc), glycosyl-phosphatidylinositol (GPI)-linked biosynthesis (provides the anchoring of proteins to membranes). Protein-associated O-GlcNAc (Figure 1.) is an O-type glycosylation, but it only adds a single O-linked β-N-acetylglucosamine molecule to the serine and threonine residues of target proteins (15). O-GlcNAc is also unique in terms that O-GlcNAc occurs mainly in the cytoplasm and nucleus. O-linked glcNAc is a reversible reaction while the remaining posttranslational glycosylations take place in the ER or Golgi and are permanent. O-GlcNAc is a fast, dynamic process; short treatment of glucosamine or exposition to a wide variety of stress conditions can lead to increased levels of protein-associated O-GlcNAc (11,16). On the other hand, after recovery from the stimuli, O-GlcNAc levels return to previous values over a relatively short period of time (11). Since O-GlcNAc needs Ser/Thr OH-residues, it can compete with phosphorylation indicating a possibility to modulate the phosphorylation-dependent signaling cascades (1,17-19). However there is also evidence that both phosphorylation sites and O-GlcNAc sites can co-exist on the same protein, influencing the protein’s function in this case in a co-operative rather than a competitive way (20, 21).
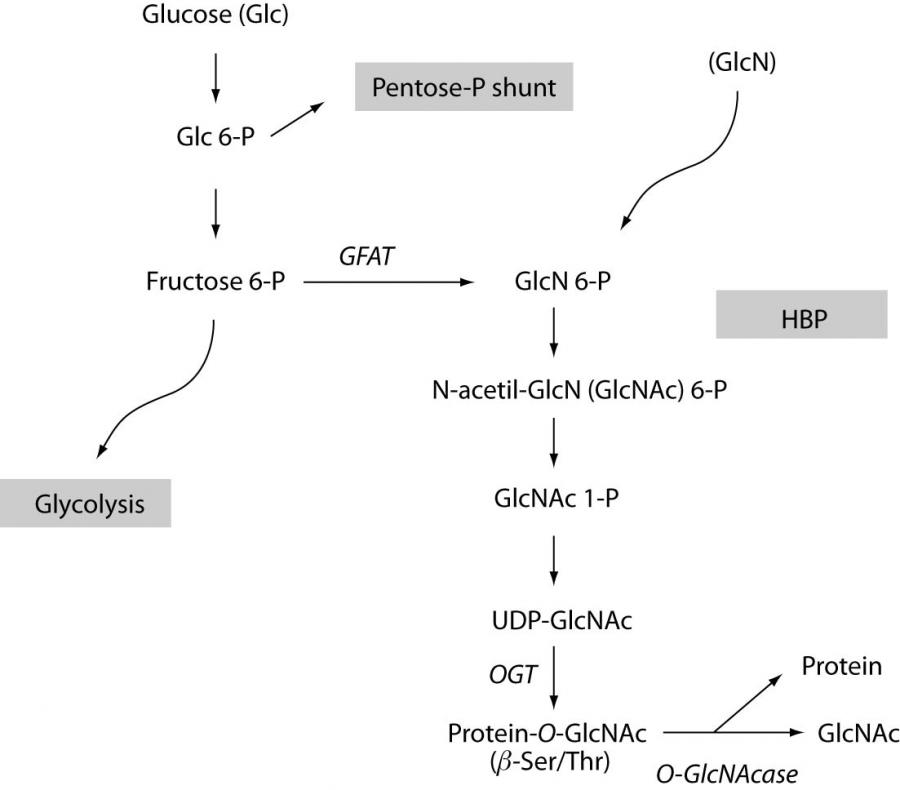
Figure 1. The hexosamine biosynthesis pathway
Interestingly, while there is an abundance of kinases, only one gene encodes O-GlcNAc transferase (OGT) which is located on the X chromosome (22). The regulation of OGT has not been well understood yet, but there is evidence that OGT itself is a target for both phosphorylation and O-GlcNAc (23). The removal of the N-acetylglucosamine group from proteins is also catalyzed by a specific enzyme called O-GlcNAcase and its gene is localized on the 10th chromosome (24). To prove that a protein in vivo is indeed O-glycosylated requires extensive, multiple approaches such as immunoprecipitation, 2D electrophoresis and mass spectrometry, so that in most studies researchers decide for the measurement of the overall O-GlcNAc levels simultaneously with the activity and expressional levels of the proteins of interest. The most commonly used specific antibodies against O-GlcNAc proteins are called CTD110.6 and RL-2 (25,26). There are multiple ways to influence O-GlcNAc levels experimentally: e.g. by overexpression or by gene deletion/gene silencing of key enzymes such as GFAT, OGT and O-GlcNAcase (11,27-29). It has to be noted that even though HBP-defected cells lines can be created, the presence of O-GlcNAc is vital and OGT-knocked-out animals die at embryonic stage (22). To simulate diabetic conditions, the flux through HBP can be increased by adding external glucosamine or high levels (25-30 mM) of sugar (29). When working with insulin-dependent cell types and with no alternate energy source available (e.g. lactate), care should be taken when cells are exposed to glucosamine for a prolonged period of time since glucosamine may deplete ATP levels (30).
It has also been reported that glutamine treatment has similar effect (through enhancing GFAT activity) (31). Azaserine and 6-diazo-5-oxonorleucine(DON) inhibit GFAT, thus decreasing the flux through HBP, while alloxan inhibits OGT, decreasing only the levels of O-GlcNAc and leaving the other metabolites of HBP relatively unchanged (32). O-(2-Acetamido-2-deoxy-D-glucopyranosylidene)amino-N-phenyl-carbamate (PUGNAc) and streptozotocin (STZ) block O-GlcNAcase and thus inhibit the removal of O-GlcNAc from proteins (33,34). It should be noted that alloxan and STZ are non-specific inhibitors; although STZ especially has a debatable effect, it is a widely used drug in animal models to induce type 1 diabetes by destroying pancreatic β-cells.
The number of identified proteins capable of posttranslational O-glycosylation is quickly growing, to date they include more than 400 cellular proteins, such as NF-κB, annexin, endothelial nitric oxide synthase, αB-crystallin, OGT, α-tubulin, c-myc, heat shock protein 70 etc. To aid O-GlcNAc research, Center for Biological Sequence Analysis web site, available at:http://www.cbs.dtu.dk/services/YinOYang/ produces neural network predictions for O-ß-GlcNAc attachment sites in eukaryotic protein sequences.
Influence of O-GlcNAc on protein function
O-GlcNAc modification of proteins definitely induces changes in the functionality of these proteins; the first and most thoroughly investigated function of O-GlcNAc is the relation to phosphorylation (35,36). Wells and co-workers have shown that OGT and protein phosphatase1 co-exist in a common complex (37). In some proteins, reduced O-GlcNAc levels correlate with increased phosphorylation levels (Tau from the brain tissue of Alzheimer’s disease patients (38)). Also, a recent report has shown p38 phosphorylation to be a subject to O-GlcNAc modification (39). It has been shown that inhibition of kinases such as PKC and PKA may increase O-GlcNAc levels (35). If these data are taken together, the most common interaction of phosphorylation and O-GlcNAc seems to be reciprocal.
O-GlcNAc may also block protein degradation, either by blocking phosphorylation sites which are required to promote the degradation (e.g. estrogen receptor (ER)-β which has a PEST sequence = The short life-time of a protein is signaled by a region rich in the amino acids proline (P); glutamic acid (E); serine (S); and threonine (T)) or O-GlcNAc directly blocks the degradation target sites of the proteins (e.g. Sp1) (40,41). There is also evidence suggesting that the proteasome is O-glycosylated and the level of O-GlcNAc on proteasomes depends on the nutritional state of cells (42). According to this hypothesis, glucose levels and consequently HBP flux and O-GlcNAc decrease during cell-starvation and thus the proteasome would be liberated from the inhibition allowing protein degradation to provide energy.
Apart from the above mentioned mechanisms, O-GlcNAc may also regulate protein-protein interactions and protein localizations (reviewed in 1, 43). As a carbohydrate, the addition of O-GlcNAc may change the hydrophobicity of a protein. To date, little is known about the effect of O-GlcNAc on protein hydrophobicity, although there is some evidence that O-GlcNAc may change the hydrophobic interactions between proteins (42). Also, our recent, yet unpublished results showed altered osmotic resistance and intracellular water diffusion following glucosamine treatment.
The role of O-GlcNAc in intracellular processes
Nutrient sensing
Strong evidence suggests that HBP and O-GlcNAc take a significant part in nutrient sensing (3). High glucose levels generate increased flux through the HBP and subsequently elevate O-GlcNAc, which downregulates glucose uptake by a negative feedback mechanism. However, not only glucose but free fatty acids, glutamine and glucosamine also increase HBP flux (3,44-46). Elevated O-GlcNAc first inhibits glucose entrance through the cell membrane by increasing the insulin resistance. The exact mechanism underlying insulin resistance is not completely understood, yet there are several studies showing that the translocation of glucose transporter GLUT4 to the cell membrane is damaged when high levels of glucose or glucosamine are administered (47,48). This phenomenon can be linked to, e.g., impaired AKT activation which is presumably required for insulin-dependent GLUT4 translocation (see below).
Glycogen synthesis is also modulated by O-GlcNAc (49). Glycogen Synthase (GS) is deactivated by Glycogen synthase kinase 3 (GSK-3). Insulin, through PI3kinase, AKT and PKC inhibits GSK-3 thus causing increased GS activity (50). One way of O-GlcNAc regulation is to block the signaling pathway induced by insulin. As mentioned, it has been reported that upon increased O-GlcNAc, AKT activity was reduced either by direct inhibition of phosphorylation or by upstream kinase inhibition (36). On the other hand, it has been shown that GS itself can be O-glycosylated, and this modification inhibits its function just like phosphorylation by GSK3 (21). The putative mechanism here is a good example to show that phosphorylation and O-GlcNAc co-operate in the same protein to block its activity.
Cell cycle
There are many O-GlcNAc susceptible proteins that are involved in cell cycle, such as the proto-oncogen c-myc (51), or cytoskeletal proteins (regulation of the mitotic spindle during cytokinesis) as α-tubulin and keratin 8, 13, 18 (52-54). YY1 (55), a protein involved in DNA replication, cell growth and differentiation is also O-GlcNAc modified. Inadequate regulation of cell cycle is a major factor in the development of cancer, so that the understanding of the role of O-GlcNAc is essential. Disruption of the HBP (e.g. by gene-deletion of glucosamine-6P-acetyltransferase) results in significantly lowered overall O-GlcNAc levels. Failing HBP is lethal for experimental animals in embryonic phase (22), yet HBP-defective cell lines can be maintained. Still, these cell lines have slower growth rates and altered cell cycles (56). Since elevated O-GlcNAc also disturbs cell growth, O-GlcNAc regulation seems to be different during the various phases of the cell cycle. Slawson et al. have recently shown that proper O-GlcNAc processing is required for normal cell cycle and that O-GlcNAc is necessary for the fine-tuning of the M-phase progression, cytokinesis, and mitotic protein phosphorylation (4).
Transcriptional factors
In general, the bulk of O-GlcNAc-modified proteins are localized in the nucleus, associated to chromatin (57). The nuclear pore complex is also abundant in O-GlcNAc (58), suggesting that O-GlcNAc modulates nuclear trafficking. However, it has been shown recently that the presence of O-GlcNAc is not necessary for pore transport (59). The most important role of O-glycosylation in the nucleus is the regulation of transcription. The list of known transcriptional factors that can be up- or downregulated by O-GlcNAc is growing every day; Whelan et al. have recently published an updated list (60). Some examples are Sp1 (41), p53 (8), CREB (5) and NF-κB (7). O-GlcNAc serves as a signal to localize a protein to the nucleus (17), and also as a modification in relation to transcriptional activity. O-GlcNAc modification can increase (p53 (8)), or decrease (CREB (5)) transcriptional activity, or both (Sp1) (41,61,62). A possible explanation for the diverse functionality is that on these proteins multiple O-GlcNAc sites could be present that are responsible for either delayed protein degradation (increased degradation results in lower activity) or the regulation (positively or negatively) of transcriptional activity. NF-κB (see below) is present in all cells and is activated upon a wide range of stimuli; stress, cytokines, free radicals, or antigens. NF-κB plays a significant role in immune response, inflammation, auto-immune diseases, diabetes, cancer, and cardiac stress response so that its O-GlcNAc modification has a special importance (7, 63,64).
Ca2+ handling
Considering the wide cellular applicability of O-GlcNAc, it is plausible that it might also interfere with intracellular Ca2+ regulation, especially in stress and ischemia/reperfusion episodes where [Ca2+]i is a crucial signal transduction element. The interaction of O-GlcNAc with phosphorylation has been well established; however, so far only indirect evidence suggests that it is also involved in [Ca2+]i homeostasis (65).
It has long been known that the so-called glucose-insulin-potassium (GIK) is beneficial for patients affected by ischemia/reperfusion (66). Moreover, in animal models, short term hyperglycemia or glucosamine treatment protects from ischemia-reperfusion induced Ca2+ -overload (67). Glucosamine also seems to inhibit Capacitative Calcium Entry (CCE) – which is an IP3 induced Ca2+-increase (16, 68). As we have shown recently, this inhibition in cardiomyocytes occurs via O-GlcNAc (16), although specific target proteins are unknown. The regulation of [Ca2+]i during ischemia/reperfusion in the heart is in itself a complex mechanism not known in every detail; however, the above mentioned indirect evidence suggests that HBP and/or O-GlcNAc modulate the [Ca2+]i homeostasis.
During an ischemic episode or upon stimuli by an an agonist such as Angiotensin II (AngII), phospholipase C (PLC) is activated and generates two secondary messengers, inositol triphosphate (IP3) and diacyl-glycerol (DAG). Ca2+ increase can be mediated by both the IP3 and the DAG/PKC route. IP3 releases Ca2+ from the ER (which is followed by a second Ca2+ influx from the extracellular space (called CCE)) while PKC/DAG activates Ca2+ channels in the cell membrane (L-type and probably other Ca2+channels as well). A number of papers has shown that the TRPC (transient receptor protein channel) membrane protein family plays an important role in the regulation of [Ca2+]i in the heart either through IP3 or PKC activation (69).
O-GlcNAc could interfere at several levels of this [Ca2+]i regulation, for example in PLC, or downstream of PLC: IP3 receptor and/or PKC and other kinases. The elimination and re-uptake of Ca2+ (Na+/Ca2+ exchangers, SERCA (sarco/endoplasmic Ca2+ -ATPase), mitochondria) might also be affected. In fact, it has recently beeen shown that PLC activity was downregulated by O-GlcNAc, suggesting that PLC is a possible O-GlcNAc target (65). TRPC proteins are also likely candidates for O-GlcNAc, e.g. the analysis of the protein sequence for TRPC1 suggests a high-affinity site for O-GlcNAc, close to the NH2-terminal region.
The O-GlcNAc-proteins concerning [Ca2+]i discussed so far affect mainly short-term posttranslational modifications. Alternatively, long term exposure to high levels of glucose also enables expressional changes since the O-GlcNAc modification of transcriptional factors can influence the expressional levels of proteins involved in [Ca2+]i handling. Indeed, SERCA2a was reported to have a decreased expression after prolonged high glucose incubation and this change was attributed to the transcriptional level (Sp1) rather than to posttranslational modification (29). Thus, even if high glucose levels can be useful for cell survival under certain circumstances, previous history of long-term high glucose exposure probably overshadows the beneficial effects in this case.
The role of O-GlcNAc in pathogenesis
Diabetes
Type 2 diabetes is characterized by increased levels of blood glucose via insulin resistance of peripheral cells, and by diabetic complications caused by prolonged exposure to high glucose. Although the exact mechanisms await further clarification, the majority of reports concur that O-GlcNAc contributes to both insulin resistance and to the development of diabetic complications.
Insulin resistance
Reduced glucose transport through cell membrane results in insulin resistance. This is caused by impaired translocation of GLUT4 glucose transporter (which is probably an O-GlcNAc protein (70)). This translocation (and also the docking and the fusion with the membrane) is regulated by multiple mechanisms, with the most important factor being insulin and insulin receptor activation. The next step involves IRS-1 and -2 (insulin receptor substrates) whose O-GlcNAc binding capacity has been well established (19,71,72). Downstream of IRS; PI3kinase, Akt, (decreased activity in insulin resistance) PKC, p38 and NF-κB (increased activity in insulin resistance) have been implicated in the insulin signaling pathway cascade. From these messengers, IRS (72), PI3kinase (19) are O-GlcNAc proteins, and putatively Akt (72), p38 (39), and NF-κB (7) are also candidates for O-glycosylation. Although the O-glycosylation of NF-κB is putative yet, the activation of NF-κB in diabetes has been well described (7). Apart from O-GlcNAc, NF-κB can be also activated by AngII or by free radicals, also contributing to insulin resistance.
The functionality of Munc18c, a regulator of docking/fusion of vesicles (containing e.g. GLUT4) to plasma membrane is disturbed when treated with glucosamine or high levels of glucose (73). Munc18c is also subject to O-glycosylation. Although all this evidence suggests that O-GlcNAc plays a major part in insulin resistance, O-GlcNAc is not by all means necessary for its development (74).
Diabetic complications
The most comprehensive hypothesis to explain the underlying molecular pathomechanism of all diabetic complications, such as accelerated atherosclerosis, peripheral nerve failure, renal and retinal complication caused by microvascular damage, was published by Brownlee (75). According to this author, hyperglycemia-induced mitochondrial superoxide overproduction blocks glyceraldehyde-3P-dehydrogenase, a key enzyme in glycolysis. Therefore the upstream metabolites are diverted to other pathways: the polyol way, the advanced end-glycation products (AGE), the activation of PKC (by increased production of DAG), and increased flux through the HBP. The HBP is associated in diabetic complications with increased expression of TGFα (transforming growth factor), TGF-β1 and PAI-1 (plasminogen activator inhibitor) (62,76,77). The exact link between TGF and HBP has not been revealed yet (PKC is assumed (78)); however, PAI-1 expression is increased by Sp1 transcriptional factor which is an O-GlcNAc protein.
Endothelial nitric oxide synthase (eNOS) is also O-glycosylated, the O-GlcNAc masking of the Akt phosphorylation sites on eNOS prevents its activation, thus decreasing the levels of the NO which is a strong vasodilatator (19,79). HBP indirectly activates also the PKC pathway, although apparently not by direct O-glycosylation of PKC itself, but probably by the involvement of other upstream kinases (80). Taken together, these pieces of evidence strongly suggest that protein O-glycosylation regulates a great number of cellular processes related to diabetes, and that the long term disturbance in HBP and/or O-GlcNAc-handling leads to severe diabetic complications.
Stress response
As mentioned above, glucosamine and high glucose prevent ischemia/reperfusion injury and Ca2+ paradox. Recently, it has been shown that selective increase in the levels of O-GlcNAc has a similar effect (14,16). Either in perfused heart or in trauma-hemorrhage rat models, glucosamine caused both increased levels of O-GlcNAc and simultaneously reduced ischemic damage (14,81). In isolated cardiomyocytes, PUGNAc, a specific inhibitor of O-GlcNAcase, also protected from hypoxic damage (82).
On the other hand, without any external intervention cells tend to increase their O-GlcNAc levels upon stress. As it was shown by Hart and coworkers, the stress response to a number of different stress (heat, hypoxia, osmotic stress) included elevated O-GlcNAc (11). Blunting OGT not only abolished the stress-elevated O-GlcNAc but reduced the stress tolerance and cell survival. This result seems to support the hypothesis that O-GlcNAc is a necessary element of normal stress response.
Which are the specific target proteins related to stress activated O-glycosylation? Heat shock proteins seem to be the first reasonable answer, indeed a number of Hsp-s are candidates for O-GlcNAc (51,83). It was also shown that the expression of Hsp70 is increased after O-GlcNAc modification (11). For O-GlcNAc, another subject of regulation could be the [Ca2+]ihomeostasis. Hypoxia or stress elevates [Ca2+]i, and Ca2+ mediates a number of deleterious effects unless the initial stress is removed quickly. Elevated [Ca2+]i activates intracellular messengers such as calcineurin, calmodulin, NF-AT, PKC and caspases. This results in the activation of several transcriptional factors and the cells end up either in hypertrophy or apoptosis. Interestingly, hypoxia induces glucose transport by Ca2+ (84) which is an indirect evidence for the link between [Ca2+]i and O-GlcNAc regulation. As mentioned above, we have shown that manipulations with O-GlcNAc influenced [Ca2+]i regulation in cardiomyocytes (16). In cardiomyocytes, elevated levels of O-GlcNAc, achieved either by increasing the flux through the HBP by glucosamine treatment or inhibiting O-GlcNAcase with PUGNAc, prevent the increase of basal [Ca2+]i induced by AngII (Figure 2). This inhibitory effect of O-GlcNAc on [Ca2+]i level develops possibly via multiple targets, e.g. PLC or TRPC channels.
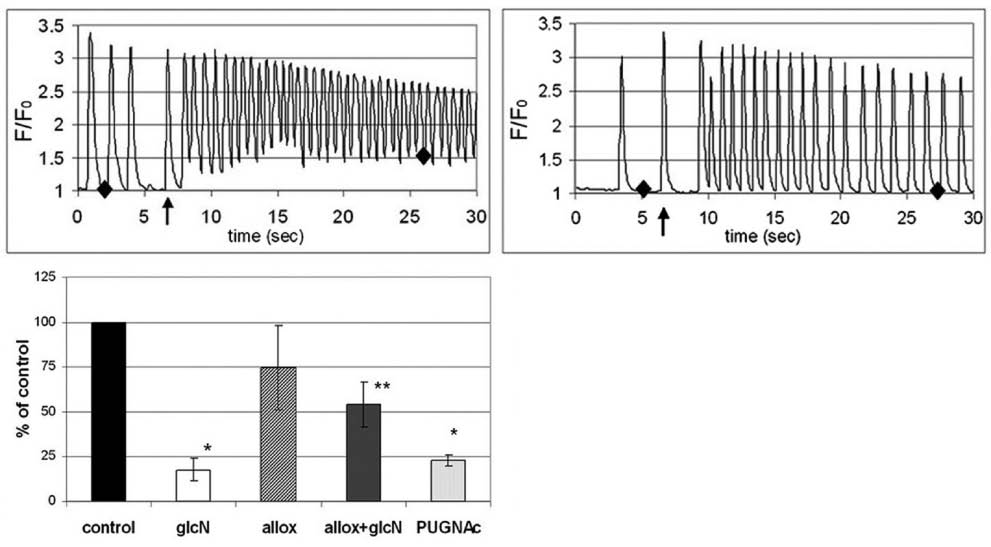
Figure 2. Increased O-GlcNAc levels inhibit AngII-induced [Ca2+]i rise. A.) Left: AngII (indicated by the arrow) treatment causes rapid increase in diastolic [Ca2+]i in neonatal rat cardiomyocytes. Right: pretreatment with 5 mM glucosamine for 10 min inhibits the diastolic [Ca2+]i increase elicited by AngII. B.) Changes in baseline [Ca2+]i after AngII treatment, relative to controls. Both glucosamine and PUGNAc, an inhibitor of O-GlcNAcase, reduced the increment of [Ca2+]i whereas alloxan, an inhibitor of OGT, partially reversed the effect of glucosamine.
The role of O-GlcNAc in stress response could be described as follows: stress activates several signaling pathways, most importantly Ca2+ which indeed at first serves as a natural and necessary adaptation mechanism. [Ca2+]i then facilitates the entering of glucose into cells, which provides additional energy source. However, a small portion of the glucose will flux through the HBP, inducing the modulation and downregulation of [Ca2+]i and stress response by O-GlcNAc. If the stress stimulus is short-term or limited, HBP can prevent the cells to over-react, however if the stimulus is prolonged (but still sub-lethal), the physiological O-GlcNAc elevation is often not sufficient to counter the deleterious effects of Ca2+-overload.
O-GlcNAc also fits well into the preconditioning theory: a short, mild ischemia or stress reduce the risk and seriousness of a subsequent ischemic attack [85]. Preconditioning was attributed to a number of signaling pathways, such as Akt/PI3kinase or PKC (86). Since stress increases O-GlcNAc levels, O-GlcNAc elevates during the first stimuli and by the second exposure it could help to reduce the stress-induced damage. As above, we assume that this effect occurs via multiple mechanisms and targets rather than by modifying a single selected messenger. It is plausible that the overall O-GlcNAc level of the cell reflects the general stress tolerance and adaptation state at a given time.
The effect of HBP and O-GlcNAc in diabetes and in stress response is quite contradictory. A possible explanation is that while diabetes is a long term, chronic disease and the associated hyperglycemia and elevated O-GlcNAc need months or years to develop diabetic complications, the stress-induced O-glycosylation is an acute, very rapid condition (12). This means either that in diabetes and in stress different proteins are modified, or that the same proteins are involved, but the initial, short term activation is not sufficient to initiate the activation of signaling cascades and the deleterious effects observed in diabetes.
It is well known that diabetic patients have a significantly increased risk for cardiovascular damage and ischemic episodes. This needs a long time exposure to a high level of glucose which irreparably damages the cardiovascular system. During an acute ischemic attack, the possible regulatory O-GlcNAc elevation is insignificant in terms of preventing hypoxia-induced injuries. On the other hand, a non-diabetic patient could better benefit from elevated glucose and HBP, as it has already been implicated by the use of GIK infusions. Although it is highly theoretical, glucosamine treatment could grant the same or better results. Glucosamine is already a widely used therapeutic drug in osteoarthritis. It may have a pro-diabetic effect, although the studies so far could not prove this unequivocally (87). It seems that it is relatively harmless at normal dose or at short term application.
Inflammation
The role of O-GlcNAc in inflammation is controversial. In diabetes, NF-κB is activated, and inflammation mediators also increase their expression, such as TGF-β1 or PAI-1 (62,77). TGF-β1 overexpression is probably related to PKC activation (78), which is implicated in insulin resistance and diabetic complications, whereas PAI-1 promoter is activated by Sp1, one of the first transcriptional factors that has been found to be O-glycosylated.
On the other hand, a couple of studies described that glucosamine treatment inhibits NF-κB in conjunctival cells (88) or in chondrocytes (13). Apparently, this inhibition could explain the beneficial effects of glucosamine in osteoarthritis. It has been also reported that glucosamine prevents CD3-induced T cell proliferation (89), and found that it prolongs cardiac allograft survival in mice (90). The authors of the latter publication propose that the anti-inflammatory and immunosuppressant effect of glucosamine might be connected to transient exposures whereas insulin resistance requires the presence of glucosamine continuously.
Malignant diseases
There is relatively scarce information available about the role of O-GlcNAc in malignant diseases. However, O-GlcNAc plays a significant role in cell cycle, and numerous transcriptional factors are subject to O-glycosylation. For example, c-myc proto-oncogene was found to be O-glycosylated and the O-GlcNAc site(s) are located within or near the N-terminal transcription activation/malignant transformation domain, a region where mutations are frequently found in Burkitt and AIDS-related lymphomas (91). The tumor suppressor p53 was also found to be O-glycosylated; O-GlcNAc seems to modulate its DNA binding capacity (8) or to block phosphorylation which delays proteolytic degradation of p53 (92).
The transcriptional factor Sp1 is also often associated with cancers (93). Correlating with hypoglycosylated state, Sp1 is rapidly degraded by the proteasome and this degradation can be prevented by glucose or glucosamine treatment (41). Second, Sp1 contains a single O-GlcNAc residue whose modification inhibits hydrophobic interactions between Sp1 and two partners, the TATA binding protein-associated factor (TAFII110) and holo-Sp1 (61). Roos et al. propose that, upon DNA binding, Sp1 has to lose its O-GlcNAc residue by a flip-flop mechanism with phosphorylation in order to bind TAFII110, holo-Sp1 and induce transcription.
The ‘hypermethylated in cancer 1’ gene (HIC1) is a candidate tumor suppressor gene, and is modified by O-GlcNAc in a number of malignant cell lines; however, the O-glycosylation seems to affect stability and not DNA binding affinity (94). Finally, RNA polymerase II can be modified by O-GlcNAc. It has been reported that OGT interacts with a histone deacetylase complex by binding to the corepressor mSin3A, and represses transcription in parallel with histone deacetylation. mSin3A targets OGT to promoters to inactivate transcriptional factors and RNA polymerase II by O-GlcNAc modification (95).
The mode in which O-GlcNAc influences the development of malignancies is still controversial, since it can prevent the degradation of transcriptional factors, but also seems to directly block or activate the same factors. Understanding the complex behavior of O-glycosylation will require taking into account the localization, spatial organization of OGT and also the mapping of the individual (and possibly multiple) O-GlcNAc sites on transcriptional factors.
Conclusions
The evidence established in the last two decades shows that O-GlcNAc is a unique, but important intracellular signaling mechanism, covering and participating in almost every cellular event, being either physiological or patho-physiological. Although O-glycosylation is a general, common process in the cell regulated by a single enzyme - OGT -, the mechanism, the effects of, and the proteins subjected to O-GlcNAc modifications are highly specific. This is achieved by spatial and timed organization, and by a harmonized coordination with phosphorylation. The modulation of a number of signaling events in diabetes, stress, malignant diseases or in inflammation undoubtedly deserves the attention in further research. Although elevated levels of O-GlcNAc are clearly detrimental in diabetes, clarifying its role in acute stress response could be a great step forward to improve the prevention of ischemia/reperfusion injuries. Hopefully the better understanding of O-GlcNAc in the future will help to both reduce diabetic complications and to increase the life expectancy of ischemic patients.