A survey of previous concepts on biological changes in mood disorders
In addition to schizophrenia, anxious disorders, alcohol and other addictions, mood disorders pose a major research challenge for both clinician psychiatrists and neuroscientists. There is ever more evidence for the biological causative factors which, together with environmental social factors lead to mental disorders (1). This interaction is very complex and imbued with an array of environmental factors that may modify the biological balance of the CNS, and vice versa, the whole system from molecules and ions, through cellular structures, neurons and glial cells to neurotransmitters may in certain macroanatomical structures of the CNS influence the expression of a personality, thus exerting an impact on the individual’s social environment (2).
In this sense, mood disorders are of special interest because by quality they represent mood deviation in the constant pattern of everybody’s personality. Mood is a long-standing, inner and predictable emotional state of the individual. Mood disorders used to be referred to as affective disorders, which should definitely be abandoned because mood, unlike affect, is a permanent quality, so the mood to affect relationship could be described as the climate to current weather relationship (2).
Generally, mood can vary toward low, depressive mood or toward high, euphoric, manic mood. Normal mood variation caused by daily living should certainly be distinguished from psychopathological ones. In other words, mood oscillations that depart from the daily living mood variation by severity, duration and impossibility to control are classified as mood disorders. These mood changes usually occur without any overt external cause.
As mentioned above, the biological, social and psychological factors all play a major role in the onset of mood disorders. Therefore, biological, psychological and social therapeutic procedures are used in the management of mood disorders (2). Currently, biological procedures, primarily psychopharmacological therapy, are the first-choice and most important approach in the management of mood disorders. There is a wide array of psychopharmaceuticals available, including antidepressants, antipsychotics, anxiolytics and mood stabilizers, to mention only the most common agents used in the treatment of mood disorders (1,2). The latter, mood stabilizers, in particular lithium as the oldest representative of the group, are of special interest. Mood stabilizers, including lithium salts, are used to treat and alleviate discomforts, and, unlike other psychopharmaceuticals and drugs in general, also for the prevention of recurrent disease episodes (3).
The biological etiological factors have been best studied to date and therefore provide most information on the causes of mood disorders. For didactic reasons, they are usually categorized as genetic, neuroendocrine, circadian and sleep-related, neurochemical and neuroanatomical factors (3).
Genetic factors
Genetic factors appear to play an important role in mood disorders (4). Evidence for the impact of hereditary factors is especially strong in case of bipolar affective disorder. Yet, it should be noted that mood disorders are not inherited as a dominant or recessive trait but most probably are polygenic diseases (4-6). In other words, a number of different genes are probably involved in the occurrence of mood disorders. Genes on the chromosomes 5, 11 and X are most frequently mentioned (7). Polymorphisms of the genes for serotonin receptors, serotonin transporter, dopamine transporter and dopamine receptors have currently been intensively studied. Initial results have produced controversial findings.
Bipolar affective disorder will occur in 25% of children with one of the parents affected with the disease. The probability for child affection rises to 50%–70% if both parents suffer from bipolar affective disorder, while the risk in monozygotic twins is as high as 90%. These rates are lower for depressive disorders. When a monozygotic twin suffers from depressive disorder, the likelihood for the disorder to develop in the other twin is 50%. If one of the parents suffers from depressive disorder, the probability of the child to develop depression is 10% (8).
Neuroendocrine indicators
As endocrine system is mostly regulated via hypothalamic-pituitary-target glands (thyroid gland, adrenal gland, gonads), i.e. the entire system is inter-related via hypothalamus and is an integral part of the CNS – limbic lobe, it is quite logical to expect that hormones may act as peripheral indicators of the neurochemical function of the brain (9,10). Indeed, pituitary hormones are primarily influenced by monoaminergic neurons. So, serotonin, norepinephrine and dopamine play crucial role in the secretion of adrenocorticotropic hormone (ACTH) and cortisol, thyroid-stimulating hormone (TSH), triiodothyronine (T3), thyroxine (T4) and prolactin (PRL) (11). The hypothalamic-pituitary-adrenal (HPA) axis is of particular interest in patients with depressive disorder. The dexamethasone suppression test is one of the most reliable indicators of mental disease in general. About 50% of depression patients show the lack of suppression on dexamethasone testing (11). In addition, almost all these patients have elevated concentrations of serum cortisol and low ACTH levels. Virtually every patient with thyroid disease, either hyperthyroidism or hypothyroidism, has some symptoms of mood disorder (9,10). On the other hand, patients with mood disorder frequently show elevated levels of free T3 and T4 hormones. Yet, in contrast to dexamethasone test, the thyrotropin-releasing hormone test (TRHt) is not so highly specific in depression patients (9,10).
Sleep disorders and circadian rhythms
Sleep disturbances are one of the major physical symptoms of mood disorder, either manic or depressive. Electroencephalography (EEG) abnormalities in sleep are very pronounced in patients with mood disorder. For example, the most common abnormality refers to the length of non-rapid eye movement (NREM) sleep and rapid eye movement (REM) sleep (12). As a rule, depressive patients have REM sleep episodes reduced in length and number. Circadian rhythms have also been intensively investigated, with seasonal affective disorders attracting high interest. The interrelationship of mood and melatonin “biological clock” has been studied (12).
Neurotransmitters
Mood disorders have also been associated to monoamine (serotonin, dopamine and norepinephrine) decrease in depression or their increase in mania (13). Some studies have confirmed these observations, whereas others report contradictory results. Furthermore, the agents used in the treatment of depression increase the concentration of monoamines in the synaptic cleft through various mechanisms (13). In mood disorders, the above mentioned neurotransmitters have been investigated in cerebral tissue, cerebrospinal fluid (CSF), plasma and urine. Monoamine metabolites such as 5-hydroxyindoleacetic acid (5-HIAA), homovanillic acid (HVA) and 3-methoxy-4-hydroxyphenylglycol (MHPG) have also been investigated. It would be too naïve to take the hypothesis on a single neurotransmitter system seriously. Namely, it is quite clear that neurotransmitter systems in the CNS behave according to the principle of communicating vessels, i.e. a deviation in one neurotransmitter system immediately entails modification in another (14) resulting in diversified findings in the studies of neurotransmitters in mood disorders.
Furthermore, if the concentrations of neurotransmitters or their metabolites are within the normal limits but there is a deficit or excess of neurotransmitter receptors, their action will be variedly manifested (15). Recent studies of the action of antidepressants show their therapeutic effect to be eventually manifested by down-regulation of monoamine receptors (15). Besides a number of classifications, for this purpose neurotransmitters can be classified into excitatory (glutamate acting via kainate and N-methyl-D-aspartate (NMDA) receptors), inhibitory receptors of γ-aminobutyric acid (GABA), and neuromodulatory that are both monoaminergic (dopamine, serotonin, norepinephrine, epinephrine) and cholinergic neurotransmitters. These neuromodulatory neurotransmitters may assume both excitatory and inhibitory action at a given moment, depending on the level and mode of glutamate stimulation. In other words, in mood disorders a decrease or increase of monoamines (or their enhanced or suppressed action) may result in mood elevation or depression (13-15). Yet, their level primarily depends on the concentration and excitatory action of glutamate via NMDA receptors or on the inhibitory GABA action (16).
It should also be noted that neuromodulatory neurotransmitters exert their action upon having bound to the cell receptor via G protein and second messenger system (16). The excitatory neurotransmitter glutamate or the inhibitory neurotransmitter GABA act upon ion channels, i.e. glutamate and GABA act immediately, whereas monoaminergic effect can also be expected in a prolonged period of time.
Neurotransmitters and signal pathways
It would be too simple to explain mental disorders or “normal” functioning exclusively by the modification of neurotransmitters or their receptors. As stated above, the result of the action of neurotransmitters is only achieved through the activation or inhibition of other messengers, which eventually change gene expression and thus also the phenotype. Figure 1 shows schematic presentation of the second messenger activation, where neurotransmitter is considered “first messenger” which acts, via its target receptor, upon G proteins found in the cell membrane.
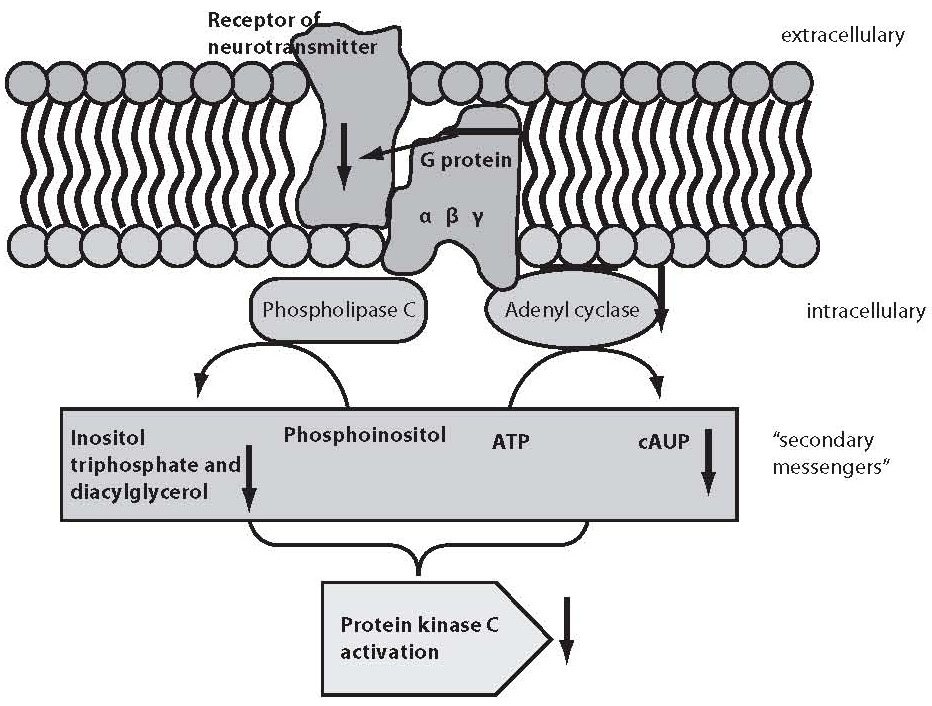
Figure 1. Schematic presentation of second messenger activation. Neurotransmitter as a “first messenger” acts upon receptor on the cell membrane, followed by the G protein and phospholipase C or adenyl cyclase mediated activation of “second messengers” that eventually stimulate protein kinase C, which then acts through a signal cascade on the early response genes.
G proteins transfer the message from neurotransmitter receptor to particular enzymes serving as “second messengers” or to ion channels. G proteins are constituted of three subunits, α, β and γ (15). G proteins act upon a series of “second messengers”, best known of which are the system of adenylate cyclase that converts adenosine triphosphate (ATP) into cyclic adenosine monophosphate (cAMP), or the phospholipase C linked system that converts inositol lipids and phosphoinositol from the cell membrane to inositol triphosphate (IP3) and diacylglycerol (DG) (13-16). IP3 and DG act upon protein kinase C or endoplasmic reticulum receptors, thus leading to the release of calcium ions to the cytoplasm. The cascade of second messenger activation is illustrated in Figure 1. It should be noted that neurotransmission and its effects are not terminated by the release of a neurotransmitter, its action upon the receptor and second messenger activation, which occurs in milliseconds (15). The final consequence of neurotransmission is the regulation of gene expression, protein synthesis, and modulation of the neuron or glial cell function and morphology according to the new conditions. These changes mostly imply strengthening or destruction as well as rearrangement of the synapses and dendrites, axon withdrawal or elongation, or even cell stimulation to death (17). The activated system of second messenger eventually activates transcription factors, which in turn activate regulatory region of the target gene stimulating the encoding gene section, thus producing ribonucleic acid (RNA) and finally the target protein (enzyme, receptor, transport protein, transcription factors, etc.) (16,17). Table 1 shows some studies of the second messenger system changes in patients with bipolar affective disorder or depressive disorder. These studies have a drawback in common, i.e. they were performed in postmortem brain samples of patients with mood disorders or on models such as platelet model. It should be noted that upon transcription factor activation, the first genes to activate are c-Fos and c-Jun, the early response genes. Upon the neurotransmitter activation of second messenger, cFos and cJun genes become active in 15 minutes and act for one hour. The products of c-Fosand c-Jun genes are Fos and Jun proteins that jointly activate or prevent activation of the late response genes (18-20). The late response genes are specific genes that regulate specific cell response to the stimulus (Fig. 2). Therefore, the entire nervous system is highly adjustable and modifiable by numerous external and inner effects; it is flexible in biochemical as well as morphological terms (20). In contrast to previous beliefs, neurons are now considered also divisible after birth. Myelinization, the number of dendrites and synapses, and axon length are changing for life, up to the neuron necrosis or apoptosis under the influence of neurotransmitters, glutamate in particular, or of hormones such as glucocorticoids under the influence of stress (21).
Table 1. Some studies of the “second messenger” system in patients with mood disorders
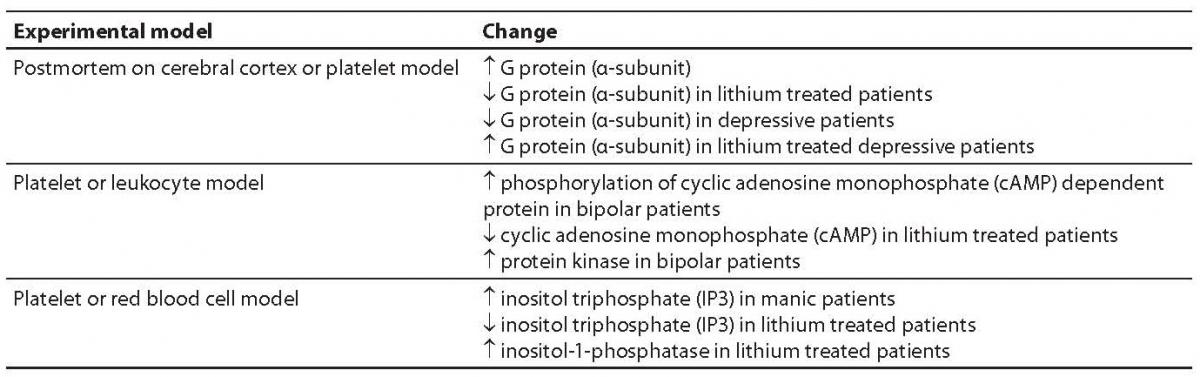
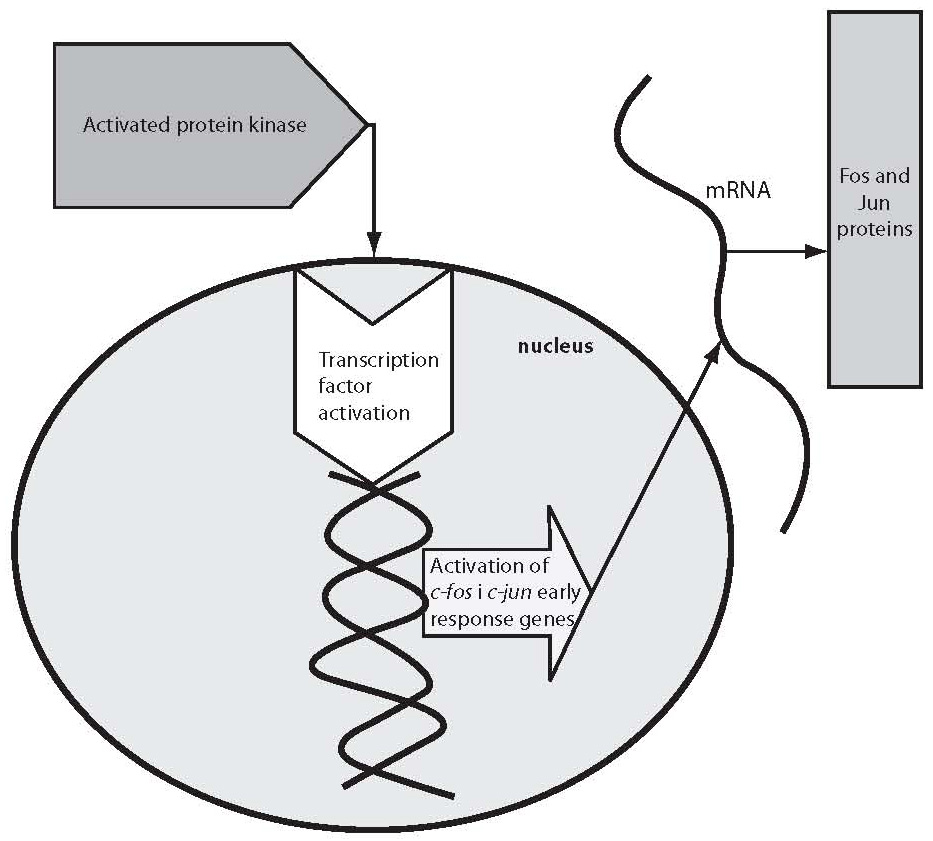
Figure 2. Activation of c-fos and c-jun genes
Neuroanatomical changes in mood disorders
Neuroanatomical investigation of mood disorders have been based on postmortem studies, morphological changes monitored by computed tomography (CT) in the late 1970s and 1980s, and nuclear magnetic resonance (NMR) studies in the 1990s. Also, mention should be made of the functional morphological methods such as single photon computerized tomography (SPECT) and positron emission tomography (PET), which have pointed to the key etiologic factors in mood disorders, e.g., diminished function of prefrontal cortex or decreased activity of the area of limbic structures, hippocampus and amygdala in particular, in depression, or contrary to this, an increased activity in the same regions in the manic phase of the disease (56-22). NMR studies also showed the frontal lobe to be smaller in patients with bipolar affective disorder than in the control group. CT studies failed to yield any significant results, in part due to the method limitations in the study of subtle morphological alterations in CNS.
In addition to the above mentioned macromorphological methods that have produced relatively modest findings in patients with mood disorders, postmortem histology methods have provided perhaps the most consistent biological findings in psychiatry in general (23). Against expectations, glial cells have surprisingly exhibited alterations (24). To date, glial cells, unlike neurons, have been considered to be of little relevance in either functional or pathophysiological terms in psychiatric disorders (25). In patients with bipolar affective disorder as well as in those with depressive disorder, glial cells were found in lower number and density, also showing size changes (26). These changes were most pronounced in prefrontal cortex, in the region where changes are well visualized by SPECT and PET, and in amygdala and hippocampus (26). The number of glial cells in prefrontal cortex was by up to 41% lower in comparison with control samples (27,28). Furthermore, in contrast to the previous opinion that glial cells serve just as supportive tissue to CNS, now they are known to possess numerous neurotransmitter receptors and to play a major role in the concentrations of serotonin, norepinephrine and dopamine in the synaptic cleft. Yet, perhaps the most important role of glial cells is the regulation of the metabolism of glutamate, the crucial excitatory neurotransmitter. Glial cells contain glutamate transporter which “collects” excess glutamate from the synaptic cleft to deposit it in glial cells as the main glutamate storage (15). As mentioned above, besides the lower number and lower density of glial cells in bipolar affective disorder and depressive disorder, an increase in the glial cell volume has also been reported (29). It is also emphasized that alterations in the frontal cortex structure in terms of changed structural plasticity occur primarily due to glial cell abnormalities. To date, studies have been based on preparations stained by Nissl method, which cannot define the exact glial cell subtype (astrocytes, oligodendrocytes and microglia) involved by pathologic alteration in mood disorders (29). It should be noted that the changes observed in bipolar affective disorder and depressive disorder differ from those in other classic neurodegenerative diseases, where gliosis is the main pathomorphological finding. Other researchers go even beyond this concept and indicate apoptosis as the key mechanism in the occurrence of these alterations in the frontal cortex of patients with mood disorder.
Apoptosis – the possible cause of neurodegenerative changes in mood disorders
Death of any cell, including neurons and glial cells, can be the result of necrosis or apoptosis. Necrosis or acute pathologic death is a process that occurs after extensive chemical, biochemical, mechanical or other similar damage. Necrosis is characterized by swelling and rupture of the cell membrane and organelles, escape of the cellular content, and consequential contamination and inflammatory changes of the surrounding tissue (31). Apoptosis is a process of physiological programmed cell death, cell suicide (32). Should a cell undergo apoptosis or necrosis, it depends on the adverse factor concentration or length of its harmful effect, on the type of damage caused by the mentioned factors, and on the cell type (33). Furthermore, apoptosis also depends on the presence or absence of the molecules inducing apoptosis. Apoptosis was first described in 1972. The term apoptosis has been derived from the Greek words apo (off) and poptosis (falling down), describing the shedding of leaves in the autumn, i.e. the normal physiological role of the programmed cell death phenomenon (34). Morphologically, apoptosis implies shrinkage and budding of the cells, the membranes are generally preserved, and the nuclei are pyknotic and fragmented. The cell divides into membrane surrounded apoptotic corpuscles, so-called Keri bodies, each of them containing various organelles. Apoptotic cells in tissues, even in vitro, are being phagocytosed by the viable adjacent cells or specialized phagocytes. In contrast to necrosis, apoptotic cell death does not induce inflammatory reaction in the surrounding tissue (35). Apoptosis has an important physiological role in the normal development and maintenance of tissue homeostasis, e.g., in morphogenetic cell death during embryogenesis, neuron selection in the formation of synapses, glial cell removal following stressful events mediated by glutamate and/or corticosteroids, in neuronal plasticity, removal of cells lacking the necessary growth factors (e.g., neuron apoptosis due to the nervous growth factor deficit), etc. (36). As stated above, apoptosis is considered one of the likely factors in the etiology of neurodegenerative process in the CNS of patients suffering from mood disorders (30). For example, in a study conducted in vivo, flow cytometry revealed a statistically significantly greater number of apoptotic cells (leukocytes) in patients with mood disorders (bipolar affective disorder and major depression) than in the control group of healthy subjects. Another, postmortem study in patients with major unipolar depression showed an abnormally high number of apoptotic cells in the CNS, hippocampus in particular. In this study, the authors also demonstrated direct association between the great number of apoptotic cells in the hippocampus and the expression of corticosteroid receptors, considering apoptosis to occur consequentially to HPA axis impairment in patients with mood disorders (37).
Molecular mechanisms of apoptosis
As noted above, apoptosis, cell suicide or programmed cell death is the key mechanism in maintaining body homeostasis (35). Apoptotic mechanisms are of special importance in the formation of nervous system during the intrauterine period as well as subsequently during the CNS development in childhood as a response to learning, and later in adult age as a response to various stimuli from the society, whereby the brain is being shaped in a plastic manner according to environmental requirements (35). Apoptosis itself is a complex molecular mechanism in which a number of intracellular pathways are intertwined, with interaction of numerous proteins, enzymes, lipids, and modification of gene expression. For didactic reasons, the mechanism of apoptosis is presented in a simplified manner and can generally be described as internal (mitochondrial pathway leading to apoptosis) or external (receptor pathway) (Figure 3). Activation of the apoptotic internal pathway occurs consequentially to intracellular homeostasis impairment (38). In this pathway, as mentioned above, mitochondria play a central role, hence it is termed mitochondrial pathway. The external pathway of apoptosis begins with ligand binding to cell membrane receptors called death receptors, hence it is termed death receptor pathway (38). Although differing in the mode of activation, these pathways are interrelated and eventually both pathways give rise to characteristic biochemical and structural cell changes characteristic of apoptosis. As mentioned above, the external pathway is activated by ligands through their binding to death receptors (38). These receptors may vary, and they belong to the receptor family of tumor necrosis factors (TNF). A number of substrates can induce activation of the external apoptotic pathway. Considering CNS functioning in physiological or pathophysiological conditions, the following substrates may significantly influence death receptors on the cell membrane: neural growth factor (NGF), brain derived nuclear factor (BDNF), interleukins, glutamate, and neurotrophins (38). Upon ligand binding to receptors, adaptive molecules and effector caspases (caspase-8 and caspase-10) are bound to the complex. This leads to the activation of procaspases, cleavage of effector caspases 3 and 7, intranuclear DNA cleavage to specific segments, so-called “ladders”, and eventually to specific structural changes. The mitochondrial pathway starts by opening of the mitochondrial membrane pores, release of cythochrome c (and other proteins) from the mitochondria, formation of apoptosomes from cytochrome c, procaspase-9 and apoptotic protease activating factor (Apaf-1), activation of effector caspases 3 and 7, and DNA cleavage (38). Members of the B cell lymphoma 2 protein family (Bcl-2) play a key role in the regulation of apoptosis and cell survival. Bcl-2 proteins include proapoptotic proteins (Bax, Bak, Bad, Bid, Bik) and antiapoptotic proteins (Bcl-2, Bcl-XL, Mcl-1) (39). These proteins may have multiple domains (Bak and Bax) or only BH-3 domain, hence termed BH3-only proteins (Bad, Noxa, PUMA). Multiple domain molecules can be activated by interaction with BH-3 proteins. Bcl-2 proteins control (directly or by binding to proapoptotic proteins) mitochondrial permeability by modulating the outer membrane pores or regulating opening and closure of the permeability pores.

Figure 3. Internal and external apoptosis pathways. External apoptosis pathway is initiated via receptors (“death receptors”) on the cell surface. Internal apoptosis pathway begins within the cell, in mitochondria.
Mitochondrial pathway can be activated by an irreversible DNA damage. In this pathway, the central role is played by the tumor suppressor gene p53 (although a p53-independent pathway also exists). The nuclear phosphoprotein p53 has a key role in the signal pathways through which the cell “feels” stress and responds to it (40). Genome stability, cell cycle and apoptosis are under the control of p53, which is out of function in 50% of tumors. The role of p53 in maintaining genome stability includes the following: (a) retaining cells in the G1/S phase of the cell cycle to hamper replication of damaged DNA and to enable repair of DNA damage; (b) stimulation of apoptosis to destroy cells with damaged DNA; and (c) interaction with other molecules involved in these processes. In apoptosis, p53 can modulate expression of apoptotic genes involved in the internal or external pathway, enhance expression of proapoptotic genes (Bax, Noxa, PUMA), diminish expression of antiapoptotic genes (Bcl-2), and stimulate expression of death receptors (such as Fas), thus stimulating apoptosis.
Caspases (cysteine aspartate specific proteases) are central executors of apoptosis (41). Inside the cells, they are found as inactive procaspases, and are activated in a proteolytic manner, by oligomerization or cleavage by other caspases. Caspases may act as initiating (caspase 2, 8, 9, 10) or effector (caspase 3 and 7) caspases. Because of their destructive action, their activity must be regulated very precisely in order to avoid unnecessary cell death. Otherwise, studies in humans have identified fourteen caspases. Among others, a substrate they act upon is the poly (ADP)-ribose polymerase (PARP) protein that undergoes cleavage under the action of caspases. Therefore, the PARP protein cleavage is also used as a marker of apoptosis. The activity of caspases is supervised by the apoptotic protein inhibitors (42), of which survivin has a major role (43). Survivin binds directly to caspases 3 and 7, thus inhibiting their activity (44). In addition, its expression depends on cell cycle, suggesting a dual role of this protein in the control of cell cycle and regulation of apoptosis (44). It should be noted that survivin is expressed during embryogenesis but not in adult individuals, with the exception of tumor cells where a significant re-expression of survivin has been recorded (43,44).
Lithium salts – old mood stabilizers with new molecular pharmacological aspects of action, with special reference to apoptosis
Lithium salts are used as mood stabilizers and are the most widely used standard psychopharmacotherapy in patients suffering from bipolar affective disorder (15). Lithium is frequently added to antidepressants for enhancement of antidepressant action in depression patients. In addition, lithium salts have an antimanic action; therefore it is also administered to patients exhibiting a manic clinical picture (15).
Lithium is a metal, atomic number 3 and atomic weight 6.94; it is member of the IA alkaline group of metals in the periodic system of elements (45). Lithium as a new chemical element was discovered in 1817 by Johan Arfwedson. As early as 1843, Alexander Ure introduced lithium in medicine as an agent for the treatment of uric diathesis, i.e. gout. Later on, in the second half of the 18thcentury, many diseases used to be explained by the impaired metabolism of uric acid. At that time, A. Trosseani, a French, and A. Haig, an Englishman, integrated the concept of uric diathesis and uric acid metabolism in the etiology of mania and depression. Hence, lithium was administered to a wide variety of patients, and as early as 1886 Carl Lange described first results on the successful treatment of depression with lithium in Denmark; in 1894, his brother Fritz reported initial results on lithium therapy for acute depression. In 1873, William Hammound reported his results on lithium therapy for mania in the United States (15,45). At the beginning of the 20th century, lithium actually turned into a “panacea” and even used to be added to drinks. In the 1930s, initial testing of lithium salts in the treatment of arterial hypertension was launched, and lithium salts were used as a substitute for sodium chloride, i.e. table salt. In 1949, first fatal cases caused by the highly toxic action of lithium were reported, and lithium was withdrawn from the market in the United States by a decision issued by the Food and Drug Administration (FDA), placing ban on its use in humans (45). At the same time, after the favorable effect of lithium in mood disorders had been forgotten for quite a long time, Jon Cade in Australia reintroduced lithium administration to manic patients refractory to other therapeutic modalities with excellent therapeutic success, however, not for long because all his patients died within several months of lithium therapy due to its toxic effects. It was only in the 1960s that lithium gradually re-entered psychiatry by the side-door, this time with regular control measurement of lithium concentration in body fluids and dose regulation according to the serum lithium finding. In 1974, lithium salts were registered again by FDA as an agent for the treatment of mood disorders (45).
The pharmacokinetics of lithium salts
Lithium salts are generally available in the form of tablets á 300 mg. Lithium is completely absorbed within 8 h, reaching peak serum level in 1 to 1.5 h of individual dose administration. Lithium half-life in serum is 15–30 h. Upon resorption in the gastrointestinal system, lithium is gradually distributed via extracellular fluid, with special affinity for thyroid tissue, bone and substantia alba. The passage of lithium across the blood-brain barrier is slow; however, once the steady state has been achieved the CSF lithium concentration is about 40% of its plasma concentration. Steady state is achieved in 5–6 days (46). A negative correlation has been observed between plasma lithium concentration and body weight. Approximately 95% of individual lithium dose is eliminated by the kidneys, i.e. in urine. About two thirds of individual lithium dose are excreted over 6–12 h in the initial phase of elimination, followed by slow excretion of the remaining dose over 10–14 days.
Tubular reabsorption of lithium mostly occurs in proximal renal tubules and is not modified by diuretics, which act upon the ascending portion of Henle’s loop and distal tubules. Lithium is excreted in human milk, therefore its use in lactation is discouraged. Lithium therapy requires close monitoring by regular determination of serum lithium concentration, which is recommended to be between 0.5 and 1.5 mM/L (47).
The mechanisms of lithium action
Until the past decade, practically nothing was known about the mechanism of lithium action, mainly because lithium is an agent that does not primarily act upon the concentration of neurotransmitters or their receptors, which until recently were the only measurable parameters in neuropsychopharmacological studies. Some information on the action of lithium could only be obtained in the last few years, with the development of laboratory methods and techniques recording changes in cell membrane and intracellular space, and the methods of molecular biology. Lithium salts exert their effect on the CNS through regulation of the second messenger system and consequential regulation of the early response genes, thus regulating neuronal plasticity. The respective studies are summarized in Figure 4 (48). Lithium influences a number of elements of the messenger pathways: (a) G proteins; (b) protein kinase (PKC) and adenylate kinase; (c) inositol triphosphate; and (d) glycogen synthetase kinase (49).
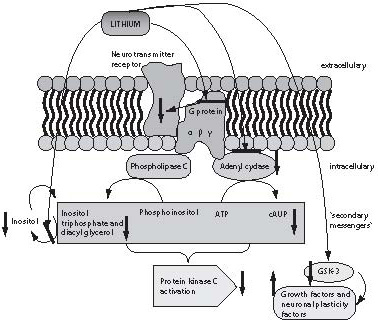
Figure 4. The mechanism of lithium action. Lithium exerts its action at several levels. Firstly, it exerts sustained action on G protein subunits modifying their conformation, resulting in G protein inactivation and inhibiting the action of neurotransmitter, which binds to the receptor. Furthermore, lithium has an inhibitory effect on adenyl cyclase and reduces the concentration of cyclic adenine monophosphate (cAMP-a) and protein kinase C. It also inhibits the enzyme for inositol triphosphate degradation, thus reducing the possibility of inositol triphosphate neogenesis (which also influences protein kinase activity). Finally, inhibition of the enzyme glycogen synthase kinase (GSH-3), a growth factor and neuronal plasticity factor inhibitor, is one of the crucial sites of lithium action.
As shown in Figure 4, lithium suppresses the action of protein kinase, which is a common substrate for both pathways of the second messenger activation, via cAMP by activation of adenylate cyclase or phospholipase C that produces diacylglycerol and inositol triphosphate from phosphoinositol. Inositol triphosphate is cleaved to inositol by inositol phosphatase. This reaction is blocked by lithium, thus reducing the possibility of inositol phosphate formation, as the inositol produced by the inositol triphosphate breakdown is reused for the production of phosphoinositol (49). The long-term effect of lithium is manifested as lower cellular excitability to neurotransmitters through conformational modification of G protein subunits, thus preventing activation of other messengers upon G protein stimulation by neurotransmitter receptors.
One of the key sites of lithium action is inhibition of the enzyme glycogen-synthase-3 (GSK-3), the crucial enzyme acting as inhibitor of many CNS cell growth factors and neuronal plasticity factors. Finally, lithium immediately induces numerous changes at the cellular level, while improvement in clinical picture is observed after days of lithium administration and action. Thus, long-term effects of lithium therapy are secondary effects that result from changes in second messengers such as transcription of genes for new proteins or growth factors, e.g., BDNF or NGF (50-52).
In our study, we found lithium administered in a therapeutically relevant concentration range to enhance expression of the survivin gene. This gene is a crucial factor in the prevention of programmed cell death. In other words, lithium enables cell survival (glioblastoma cells in this experimental model) by changing the gene expression via molecular-biochemical mechanisms (53). Furthermore, we demonstrated that 72-h lithium pretreatment prevented the antiapoptotic gene survivin expression decrease following subsequent glutamate treatment. Thus, lithium salts could also be postulated to act in mood stabilization and to prevent recurrent episodes of mood alteration. In line with numerous literature reports, we confirmed that lithium in therapeutically relevant concentration had no cytotoxic effect, in this case on glioblastoma cells (54).
Only several studies investigating the effect of lithium salts on the expression of proapoptotic or antiapoptotic genes have been reported to date. The cells of neuroblastoma or cerebellar granulated cells as cell culture were used as in vitro experimental models (55-65). Other studies were of in vivo design in experimental animals, mostly mice (60-65). The studies published so far were so designed as to observe the effect of lithium on gene expression, or the cytotoxic effect of lithium following lithium only therapy, or the effect of lithium following apoptotic mechanism stimulation with some other agent (55-65).
The effect of lithium on the cells of hippocampus was investigated by Chen et al. (55). The study was performed on mice treated with lithium carbonate solution for two weeks. Lithium concentration in mouse serum was around 1 mM (the mice were administered 2.4 g/kg lithium carbonate per day). Upon treatment, the animals were sacrificed and hippocampus specimens were submitted to immunohistochemistry and microscopy, or a part of protein was isolated from hippocampal cells to determine the content of bcl-2 by Western blot. Morphological analysis of the hippocampus showed the lithium treated animals to have a greater volume of the hippocampus, higher density of hippocampus cells, and greater number of hippocampus cells than the control specimens from animals not administered lithium. In addition, the hippocampal cells of lithium treated animals showed a higher concentration of the antiapoptotic bcl-2 gene in comparison with control, untreated specimens.
Another study performed in experimental animals investigated the effect of lithium on the cells of frontal cortex (56). The effect of lithium on the antiapoptotic bcl-2 gene expression was assessed. Like the previous study, lithium administration in concentrations of about 1 mM for two weeks led to an increase in the number and density of cells, in this case frontal cortex cells. An increase in the expression of the antiapoptotic bcl-2 gene was also recorded in the cells of frontal cortex.
Li and El-Mallahk investigated the effect of lithium on the expression of the apoptotic protein caspase-3 on neuroblastoma cells (57). This study was focused on the possible protective effect of lithium. Lithium has been demonstrated to decrease caspase-3 expression following apoptosis induced by various cytostatics. In this experimental model, lithium did not modify the expression of caspase-3, probably due to different experimental design and cell lines used. The conclusion reached in the study by Li and El-Mallahk is that lithium has a neuroprotective action and prevents increase in the expression of apoptotic genes (57).
In their study on neuroblastoma cells, Hennon et al. investigated the neuroprotective and antiapoptotic effect of lithium following ouabain induced apoptosis. Their results also pointed to the neuroprotective and antiapoptotic properties of lithium (58).
One of the interesting and properly designed studies of the effect of lithium on molecular mechanisms of apoptosis is the study by Chen and Chuang, demonstrating that lithium enhances the antiapoptotic Bcl-2 gene expression and suppresses the p53 gene expression (59). In the first part of the study, primary cultures of the mouse cerebral cells were treated with lithium at a concentration of 0.5 to 5.0 mM. In the second part of the study, after pretreatment with glutamate, which increased the expression of p53 and Bax genes, and decreased the expression of Bcl-2 gene, the cells were treated with lithium, which was found to mitigate the changes induced by glutamate (59).
In Spain, Moro et al. investigated the prevention of apoptosis with lithium and valproate as mood stabilizers following induction of apoptosis with a low concentration of potassium (60). The study was performed on primary cultures of mouse cerebral granulated cells, and was focused on the effect of lithium and valproate on phosphoinositol and protein kinase, which are increased by valproate but not by lithium. The authors conclude that both valproate and lithium exert a neuroprotective and antiapoptotic effect but via different mechanisms of action. In other words, the antiapoptotic effect of lithium is most likely accomplished by its acting upon antiapoptotic genes. In their next study, the same authors extended their investigation, demonstrating on the same cell cultures that lithium induced caspase-3 activation (61).
In their study on mouse cerebral granulated cells, Nonaka et al. investigated the effect of lithium on apoptosis induced by the antiepileptics phenytoin and carbamazepine. Using the MTT method, they demonstrated a higher survival rate of lithium pretreated cells. In addition, the mentioned antiepileptics caused apoptosis on cerebral granulated cells, with significant DNA fragmentation that could not be prevented by lithium (62).
Besides these in vitro studies, there is one in vivo study in which mice were fed standard laboratory chow and lithium salts (63). In contrast to the group of mice fed aluminum preparations alone, which have strong neurotoxic effects, the animals fed lithium in addition to aluminum showed less apoptotic cells in the hippocampus, less suppressed expression of the antiapoptotic Bcl-2 protein, and less increase in the expression of the apoptotic Bax protein (55-65).
All these literature data clearly indicate that the investigation of the molecular action of lithium has been initiated in the past few years. Even more recent are data attempting to explain the long-term effects of lithium in various experimental models, and to eventually associate, by the very complex cascade of signal pathways, the initial biochemical changes caused by lithium with mood alterations in the treated patients. Prevention of the induction of apoptosis in glial or neuronal cells, which is one of the possible pathophysiological mechanisms underlying mood disorders, has been postulated as a possible association between the action of lithium and mood stabilization (29-31). In the few studies investigating the possible antiapoptotic effect of lithium on the apoptosis induced by various stimuli, the expression of several apoptotic genes, Bcl-2, p53, caspase-3 and Bax was observed (66-70). However, apoptosis is a very complex and precisely regulated process in which numerous genes are involved (32-36), thus there probably are some other apoptotic genes the expression of which could be influenced by lithium. Therefore, additional studies are needed to confirm these mechanisms in in vivo experiments.