Introduction
Chronic myeloid leukemia (CML) is a myeloproliferative disease characterized by chromosomal translocation t(9;22) (q34;q11), also known as Philadelphia chromosome. The consequence of this translocation is a bcr-abl oncogenic fusion gene which produces bcr-abl protein with enhanced tyrosine kinase activity. Increased knowledge about CML and the discovery of the molecular pathways of tyrosine kinase has led to the creation of a “smart drug”, imatinib mesylate (IM), which competitively binds at the ATP binding site and specifically inactivates bcr-abl kinase (1,2). The introduction of IM in the treatment of CML has enabled high rate of cytogenetic and molecular remission in most patients (1). Standard treatment of chronic phase CML is 400 mg IM per day with cytogenetic monitoring of percentage of Philadelphia chromosome positive cells and molecular monitoring of bcr-abl transcript level by real time quantitative PCR (RQ-PCR) (1,3,4).
However, a proportion of patients fail to respond to IM therapy or lose the response. One of the possible reasons for that are ABL kinase mutations that are found in more than 50% IM resistant CML patients (1).
There are several types of mutations that can be divided in four groups based on crystallographic structure of cABL: mutations that bind to the catalytic domain of the bcr-abl protein and so directly impair IM, those that are within the ATP binding site, those within activation loop that prevent the kinase from achieving the inactive conformation required for IM binding, and those within the catalytic domain (1).
So far, at least 73 different mutations have been described in IM resistant CML patients (5). Not all mutations have the same relevance; some are even functionally irrelevant (3). According to response to IM they are divided into highly resistant kinase domain mutations (T315I, Y253H/K, E255K/V, H396P/R) and moderately resistant mutations (M244V, M351T, F359V). Moderately resistant kinase domain mutations can be overcome by an increase in IM dose to 600 or 800 mg per day (1,3).
The development of second generation tyrosine kinase inhibitors (TKI), nilotinib and dasatinib, that are structurally different from IM brought benefits for patients bearing highly resistant mutations because both drugs are active against all known imatinib resistant mutations except the T315I (2,5). In this mutation, single nucleotide C-->T change results in threonine to isoleucine substitution at position 315 (Thr315-->Ile) of cABL. Thr315 forms critical hydrogen bond with IM or other TKI, making the drug effective. Isoleucine contains an extra hydrocarbon group in the side chain, which results in steric clash with TKI but do not interfere with ATP binding (6). Therefore, T315I represents kind of a “gatekeeper” that controls access of small molecule inhibitors and this is why it is such a difficult mutation to inhibit, not only by IM, but also by other ATP competitors (7). Because of all the problems related to T315I, the treatment of choice for T315I carriers is allogenic stem-cell transplantation (1).
The aim of this study was to detect if the presence of T315I in patients resistant to imatinib mesylate therapy is associated with the increase or constantly high bcr-abl level. We also aimed to assess the possible difference in bcr-abl level in imatinib-resistant patients with and without T315I mutation.
Materials and methods
Subjects
The study included a total of 24 patients (10 women and 14 men) diagnosed with CML whose response to therapy with IM was inadequate. Patients were diagnosed and treated at Hematology Division, Department of Medicine, Zagreb Clinical Hospital Center. Inadequate response can be divided in three groups according to Baccarani et al. (3):
1. Suboptimal response - less than major molecular response (MMoR is defined as bcr-abl/abl level ≤ 0.1 to International Scale (IS) at 18 months of follow-up) or loss of MMoR.
2. IM failure - less than complete cytogenetic response (CCyR is defined as no Ph positive metaphase found at 18 months of follow-up), loss of CCyR or complete hematological response.
3. More than 1 log increase in bcr-abl/abl level between 2 measurements during follow-up.
At the time of investigation all patients were in chronic phase of the disease.
Methods
RNA and DNA were isolated from bone marrow or peripheral blood cells. Genomic DNA was extracted using the salting out method (8). Allele specific oligonucleotide (ASO-PCR) for T315I detection was performed according to Kang et al. (9). DNA sample from patient with confirmed T315I mutation was used as positive control.
RNA was isolated using Trizol RNA extraction protocol (10) and reversely transcribed using High capacity cDNA reverse Transcription Kit with RNAse Inhibitor (Applied Biosystems, Foster City, California, USA) according to manufacturer's instructions. RQ-PCR was performed according to Europe Against Cancer protocol (11) using TaqMan technology (LightCycler, Roche, Mannheim, Germany). Amplification of fusion bcr-abl and abl as a control gene was performed using FusionQuant Kit for RQ-PCR analysis of bcr-abl Mbcr (Ipsogen, Marseille, France) according to manufacturer’s instructions. Bcr-abl/abl levels were calculated according to IS.
Statistical analysis
Data are presented as a median and ranges and assessed by non-parametric statistics. Difference between two groups (with and without mutation) was tested with Mann-Whitney test whereas difference in bcr-abl/abl levels between three studied groups with inadequate IM response was tested with Kruskal-Wallis test. The level of significance was set at 0.05. Statistical analysis was performed using the MedCalc software package version 9.3.2.0 (Frank Schoonjans, Mariakerke, Belgium).
Results
In twenty-four CML patients who were monitored by RQ-PCR and who had inadequate response to IM therapy T315I mutation detection was performed. Electrophoretic separation of representative ASO-PCR amplification products is shown in Figure 1.
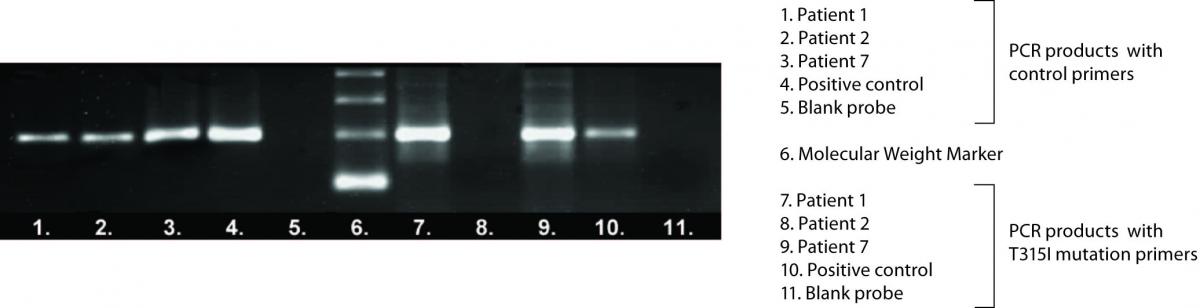
Figure 1. Electrophoretic separation of representative ASO-PCR amplification products.
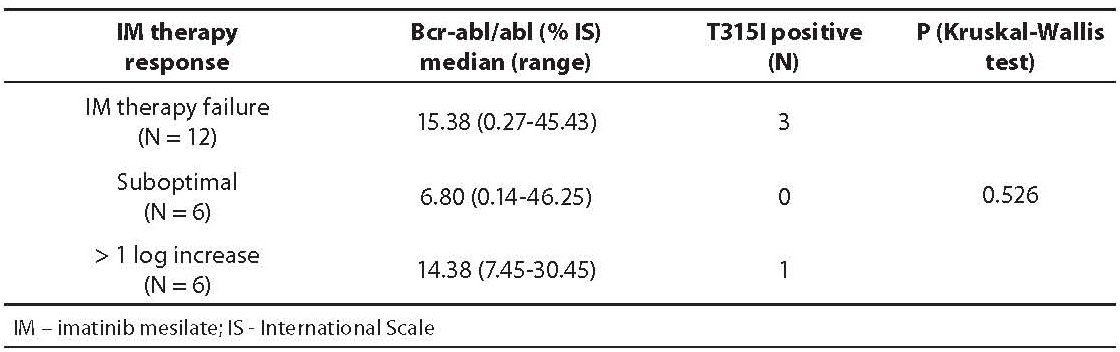
RQ-PCR results and T315I mutation status in inadequate IM therapy response groups are shown in Table 1. Among 24 CML patients studied, 12 patients had IM therapy failure (cytogenetic data not shown), 6 patients had suboptimal response, and 6 patients had more than 1 log increase of bcr-abl/abl level between two measurements during follow-up. In patients without T315I the range of bcr-abl/abl was 0.14-46.25%, unlike in those with T315I whose bcr-abl/abl range was 10.95-30.45%.
Table 1. Bcr-abl/abl levels (median and range) and T315I mutation status in three studied groups with inadequate IM response
In order to establish the possible difference in bcr-abl/abl level between CML patients with and without T315I mutation, median bcr-abl/abl levels were calculated. Results are presented in Table 2 and in Figure 2. In T315I positive patients the median bcr-abl/abl level was 66% higher than in T315I negative patients.
Table 2. Bcr-abl/abl levels (median and range) in T315I postive and negative patients
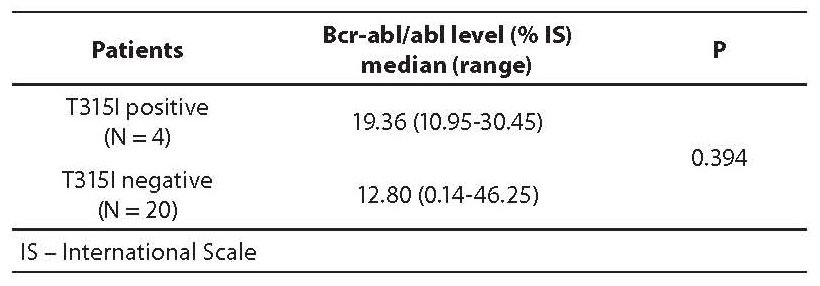
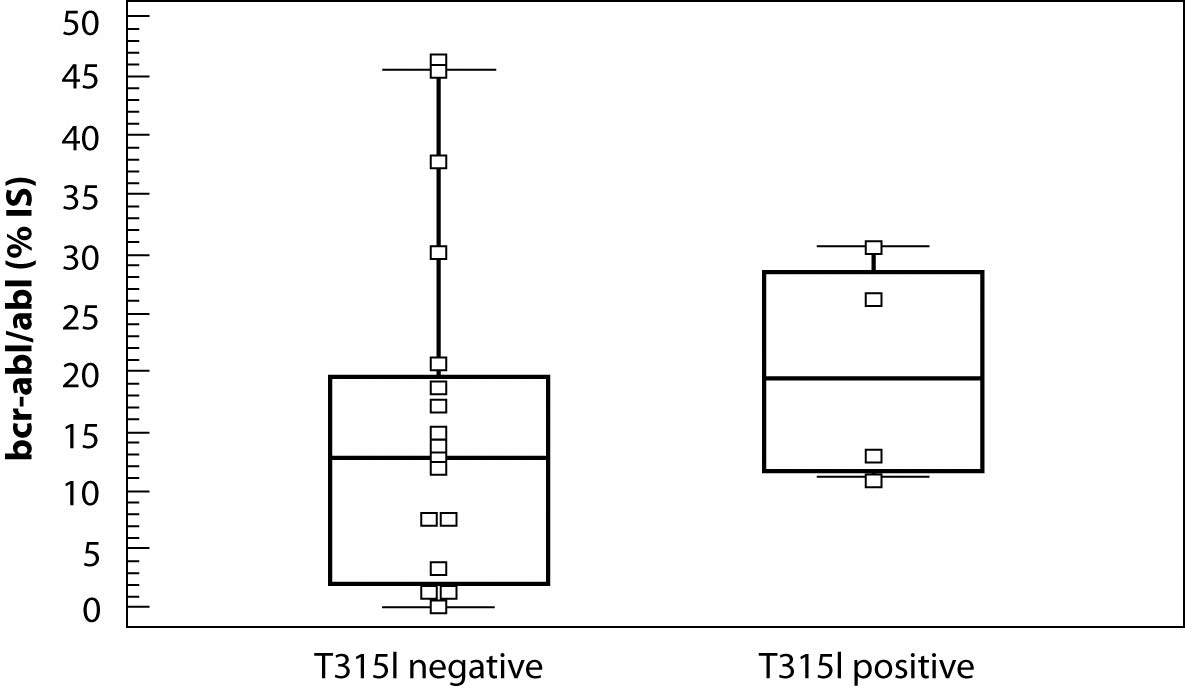
Figure 2. Box-and-Whisker plots of bcr-abl/abl levels for T315I negative and positive patients.
Discussion
After the initial description of CML more than 150 years ago, little progress has been made in the treatment of this disease for more than a century compared to the past decade. Recognition of tyrosine kinase activity of the bcr-abl proteins has led to the discovery of a new series of compounds, namely tyrosine kinase inhibitors. One of them, imatinib mesylate, was found to have high and relatively specific biochemical activity and an acceptable pharmacokinetic and toxic profile, and was rapidly introduced into clinical practice (6). This resulted in revolutionary step in CML therapy, response to therapy and consequently estimation of response by more sensitive techniques. Therefore European Leukemia Net established a protocol for CML management which includes clinical assessment, blood count, cytogenetic revision of bone marrow, and molecular monitoring of bcr-abl level every 3 months from the beginning of the therapy (3).
Major cause of resistance to IM or relapse during IM therapy are ABL kinase point mutations (7,12,13). The only mutation resistant to currently available TKI is T315I, and T315I carriers are eligible for allogeneic stem-cell transplantation only. This is why the detection of ABL kinase mutations is very useful for rational therapeutic management of CML patients (12).
Several methods have been established for detection of kinase domain mutations. Direct sequencing is one of the most used screening methods in routine monitoring of patients, but its disadvantage is low sensitivity (20%). Denaturing high performance liquid chromatography is a more sensitive method (1-5%) compared to direct sequencing. Unlike those methods, ASO-PCR has the highest sensitivity that can even reach 0.1% but the screening through the whole ABL gene domain is not possible as this method requires different primers for each mutation (1,2,12,13).
Twenty-four patients with inadequate response to IM therapy and constant increase or high bcr-abl/abl levels during follow-up were included in this study. It was assumed that, according to literature, T315I could be the possible cause (1). Among all patients tested, T315I was detected in four cases (17%). Three patients bearing T315I mutation were in IM failure group, which was expected since this mutant subclone is uniformely resistant to treatment with IM (4). In one T315I positive patient with more than 1 log increase of bcr-abl/abl level, the mutation probably expanded as a result of selective pressure exerted by IM (4).
Calculated median bcr-abl/abl levels were 19.36% for T315I positive and 12.80% for T315I negative patients, but the difference was not statistically significant (P = 0.394). These high bcr-abl levels are related to poor prognosis including progression to blast crisis and therefore indication for T315I mutation detection (3). If T315I is detected, therapy approach and treatment drastically change: patient is no more on expensive and inadequate therapy and there is more time for finding a donor for allogenetic stem-cell transplantation. Therefore monitoring is not only important for ensuring that a patient receives the best treatment, but that is also convenient from a pharmacoeconomic point of view (13).
In conclusion, T315I detection should be included in management of CML patients together with molecular, cytogenetic and clinical monitoring. The result of this analysis directly affects the therapy approach, since the treatment of choice for T315I carriers is allogeneic stem-cell transplantation.