Introduction
“Do the right thing” and “Do the thing right” (to borrow from some colleagues (1)), are two simple statements that identify the fundamental aspects of what ‘quality’ should represent to us. One particular publication (2) from the Clinical Laboratory Standards Institute (CLSI) (previously known as the National Committee for Clinical Laboratory Standards (NCCLS)) identifies several major activities of importance to a laboratory quality program, including personnel development, equipment choice and operation, process improvement, and safety. Also listed are two other essential elements that are the primary focus of the current report, namely internal quality control (IQC) and external quality assurance (EQA) for individual test processes. In addition to IQC and EQA, the current report also briefly mentions extra-analytical issues and reflects on some key guidelines available from various expert bodies to ensure performance of optimal testing within the field of haemostasis. The development of these critical elements helps to ensure that the data reported by the laboratory are as accurate as possible and that they best serve the needs of both patients and clinicians (3–7).
Overview of coagulation and haemostasis
Haemostasis, in vivo, is the homeostatic process whereby blood flow is maintained within the vasculature and, in vitro, represents one of the most complex areas of diagnostic laboratory testing (8). The main function of haemostasis, to maintain blood flow in the intact vessels in normal circulation, is in part achieved by efforts to arrest blood loss resulting from injury or trauma. There are three main phases of haemostasis, the vascular phase (which incorporates platelet interaction), the coagulation phase (which involves coagulation proteins and phospholipid complexes), and the natural anticoagulant/fibrinolytic phase (which ensures cessation of the clotting process, the dissolution of the thrombosis, and permits later wound healing). These components work in concert. When any phase of this process is disturbed, adverse clinical events may occur, manifesting as either thrombosis (haemostasis pushed in a procoagulant direction) or bleeding (ineffective haemostasis). In either situation, the clinician must have access to a variety of relevant tests in order to achieve a specific or correct diagnosis and initiate appropriate patient treatment or management.
Workers within haemostasis often refer to the above systems in terms of ‘primary’ and ‘secondary’ haemostasis, both of which promote eventual clot formation. Primary haemostasis refers to the process that involves platelets adhering to each other and to the damaged vasculature subsequent to the initiating event (e.g., tissue injury), followed by platelet activation and aggregation to produce a ‘platelet-plug’. Secondary haemostasis involves the activation of the clotting system in a complex interactive fashion with positive and negative feedback, that causes sequential activation of multiple coagulation factors and the eventual conversion of ‘soluble’ fibrinogen (the main clottable protein in plasma) to ‘insoluble’ fibrin. The process of secondary haemostasis in part acts to strengthen the platelet plug, forming a gel-like clot. This process is kept in check, in part, by the naturally occurring anticoagulant system, e.g., protein C (PC), protein S (PS), antithrombin (AT). The fibrinolytic system represents a distinct, but inter-related process that is involved in lysis of the clot to facilitate wound healing.
Common disorders and diseases of haemostasis
Many clinically relevant disease processes can be defined by a process of ‘aberrant’ haemostasis, and some of the most important include haemophilia, von Willebrand disease (VWD) and a variety of thrombophilic disorders, causing either arterial or venous thrombosis. These disorders of haemostasis can be either inherited (i.e., congenital or genetic defect) or acquired (i.e., secondary to some other disease). In order to screen, diagnose and monitor these disorders, the modern haemostasis laboratory must be equipped with an arsenal of tests to allow accurate and complete diagnoses.
Bleeding disorders
Haemophilia is caused by a deficiency of usually one and rarely several clotting factors, which may arise congenitally or may be acquired. Haemophilia A is the most common form (9) and is attributable to a deficiency or defect in factor VIII (FVIII). Haemophilia B is less common and attributable to a deficiency or defect in factor IX (FIX; also called Christmas factor). As both FVIII and FIX are essential for the process of secondary haemostasis, deficiencies of either are associated with a bleeding diathesis. Haemophilia A is approximately seven times more common than Haemophilia B. As congenital disorders, both reflect sex-linked recessive disorders as the defective genes are carried on the X chromosome. The degree of the factor deficiency can vary from a complete deficiency to upwards of around 40% of normal, with increasing bleeding severity associated to lower levels of functional clotting factors.
VWD is another bleeding disorder that arises from deficiencies or defects in the plasma protein von Willebrand factor (VWF) (10). It usually presents with mild bleeding tendencies such as epitasis and easy bruising and most often has a congenital nature but may alternatively be acquired. VWF is stored primarily in the platelets and endothelial cells and has two main functions within haemostasis (8). Firstly, VWF mediates the formation of the platelet haemostatic plug by acting as the main adhesive protein that bridges platelets to the damaged vasculature as well as to other platelets. Secondly, VWF acts as a protective carrier protein for FVIII in plasma, preventing its premature degradation. VWF functions in this manner by means of specific interactions with extracellular matrix components such as collagen, specific interactions with various platelet receptors, and a specific interaction with FVIII (8,10). VWF is thus essential for both the initial tethering of platelets to the damaged vasculature (i.e., primary haemostasis), as well as the eventual fibrin clot formation (i.e., secondary haemostasis), because it assembles with the procoagulant FVIII in plasma, forming a protective complex that prevents its rapid clearance in circulation. In plasma, VWF comprises a series of dimer proteins of increasing size (or molecular weight); the larger the size (or molecular weight), the more adhesive the molecule. Defects in VWD may comprise loss of all forms of the VWF protein (i.e., quantitative defects; defining Type 1 or 3 VWD) or selective loss of only the high molecular weight (HMW) forms or other selective qualitative defects (i.e., defining Type 2 VWD).
A host of platelet defects can also lead to mucocutaneous bleeding or bruising (11–14). Platelets constitute a store-house of haemostasis, including surface glycoprotein receptors and alpha and beta granule components. In order for platelets to exert their important role in haemostasis, these receptors need to function, and the granule contents need to be normal and released into the surrounding milieu. Platelet defects associated with bleeding often represent defects of deficiencies of platelet glycoproteins, secretion disorders, storage pool disorders, and signal transduction disorders.
Thrombotic disorders
Thrombophilia can be defined as an increased risk of thrombosis and can be hereditary or acquired. Laboratory testing of thrombophilia requires a panel of test procedures that can be molecular-based, functional or antigenic in nature. A wide variety of disorders can give rise to a thrombophilic tendency, including deficiencies or defects in the natural anticoagulant proteins (i.e., AT, PC, PS), increases in procoagulant proteins or impaired fibrinolysis (8,15–17). Thus, whilst a lack of procoagulant proteins including FVIII and VWF may cause bleeding, an excess of these may cause thrombosis. Acquired predisposition to thrombosis can also occur in many disease states, and one of the most important and common is the auto-immune disorder called the antiphospholipid antibody syndrome (APS), which arises due to the presence of antiphospholipid antibodies (aPL), detectable in the laboratory using a variety of tests including those for Lupus Anticoagulant (LA), anticardiolipin antibody (aCL) and anti-Beta2-glycoprotein-I (aB2GPI) antibodies (18,19).
Tests and assays within haemostasis
The modern haemostasis laboratory performs an arsenal of tests to allow an accurate and comprehensive diagnosis of haemostatic defects. Some tests are also used to monitor anticoagulant therapy. It is convenient to separate these tests into ‘routine coagulation tests’, which are typically high throughput screening tests performed by most clinical laboratories, and ‘diagnostic haemostasis assays’. Also commonly called ‘special coagulation’ tests, these latter assays are typically lower through-put and performed by specialized haemostasis laboratories for the specific diagnosis and characterization of haemostatic diseases and defects.
Routine coagulation tests
Prothrombin time (PT)
First proposed by Quick in 1935 (20), the PT is perhaps the oldest test of coagulation still performed. The PT is a measure of the function of the tissue factor pathway (also sometimes called the ‘extrinsic pathway’). The test is performed by adding tissue factor and phospholipids to plasma and then measuring the time in seconds to clot formation following the addition of calcium. The PT is an in vitro test that basically mimics the in vivo event of damaged tissue extracts acting on the coagulation system to initiate the clotting process. Originally developed to measure prothrombin (one of the procoagulant factors and hence its name), the PT is also now known to measure other aspects of the coagulation pathway such as the interaction of FV, FVII, FX and the concentration and function of fibrinogen.
International Normalized Ratio (INR)
This is simply a mathematical processing of the PT that permits standardization of test results across different laboratories, and is calculated by means of two additional components called the International Sensitivity Index (ISI) and the Mean Normal Prothrombin Time (MNPT) (21,22). The INR is used internationally for the monitoring of vitamin K antagonist (VKA) therapy (e.g., Warfarin), often given to individuals as a management therapy for thrombosis.
Activated partial thromboplastin time (APTT)
This test screens for abnormalities of the ‘contact-factor’ (sometimes also called the ‘intrinsic’) pathway (i.e., deficiencies or defects in FVIII, FIX, FXI, and FXII), as well as the common pathway (i.e., fibrinogen, FII, FV, FX). The APTT is also used for monitoring of heparin therapy and is variously sensitive to several inhibitors including LA (23). In all the above cases, one expects to see a prolongation in the APTT. In contrast, a shortened APTT is associated with an increased thrombosis risk (23–25). The activator in this test can be from several sources. Originally kaolin was used (hence, the test was previously called PTTK) but in more recent times activators such as micronized silica and ellagic acid have been substituted. The APTT is the time to clot in seconds when contact activator and phospholipids are added to plasma in the presence of calcium.
Thrombin time (TT)
This is a simple test performed by adding equal volumes of plasma and thrombin and measuring the amount of time until a clot forms. The TT is sensitive to heparin, direct thrombin inhibitors and to defects and deficiencies of fibrinogen. Thus, a prolonged TT will occur with congenital abnormalities such as afibrinogenaemia and dysfibrinogenaemia as well as with the presence of inhibitory substances including heparin or high levels of fibrin(ogen) degradation products (FDPs).
Fibrinogen
This represents the major clottable protein within plasma, and was originally measured by heat precipitation. These days, fibrinogen is measured immunologically or more commonly using a functional von Clauss assay, which can be easily automated. Fibrinogen levels can also be estimated using a derived calculation from the PT with selective instrumentation. The measurement of fibrinogen is important within the context of potential congenital abnormalities such as afibrinogenaemia and dysfibrinogenaemia (perhaps strangely, these may be associated with both bleeding and thrombosis) (26,27), as well as in the context of trauma, snake bite, and disseminated intravascular coagulation (DIC) (28).
D-dimer
This represents a specific breakdown fragment of fibrin, and so is a measure of fibrinolysis.This test is important in order to assess the possibility of clot dissolution as a marker of thrombosis; e.g., deep vein thrombosis (DVT) or pulmonary embolism (PE) or aberrant clot activation processes such as DIC (28–30).
Diagnostic haemostasis assays
Factor assays
These are performed by modifications of PT or APTT assays, using several plasma dilutions, in order to pinpoint specific factor deficiencies (e.g., FVIII or FIX in classical haemophilia). Tests are made individually specific for any given factor by using the particular factor deficient plasma. The exception here is FXIII, or the fibrin stabilising factor, which is measured using alternate techniques (31).
VWF assays
These comprise a panel of tests, primarily to help evaluate for a bleeding disorder such as VWD (32), although VWF is also being increasingly requested within the context of thrombotic disease (16). Some assays measure the quantity of VWF present in plasma (i.e., VWF antigen (VWF:Ag)), regardless of the presenting quality (or VWF function). Other assays measure functional properties of VWF; these include the ristocetin co-factor (VWF:RCo) assay, which assesses the ability of VWF to bind to platelets in the presence of ristocetin, and the collagen binding (VWF:CB) assay, which assesses the ability of VWF to bind to sub-endothelial matrix proteins (in this case collagen) (33). Depending on the VWF assay, a variety of techniques may be used; including enzyme-linked immunoabsorbent assays (ELISAs), latex immunoassays (LIAs), and platelet agglutination assays. Several assay limitations are evident with these tests, including assay reproducibility (34) and lower limit of sensitivity (35) issues. The VWF:CB tends to be more sensitive to HMW VWF, also suffers less variability than the VWF:RCo, and has better lower limit of sensitivity properties. Nonetheless, as the VWF:CB reflects a different functional property of VWF than the VWF:RCo, both tests are required to identify all possible forms of VWD (32,33). Several other highly specialized procedures are available to help functionally characterize or classify VWD (32,36–38), including the ristocetin induced platelet aggregation (RIPA) procedure (typically performed as a part of platelet function testing), VWF:multimer analysis (to help assess for structural defects arising from altered VWF assembly or proteolysis) and VWF:FVIII binding (VWF:FVIIIB).
Platelet function testing
This can be achieved using comprehensive platelet aggregation testing (11–14) or using a screening test process (e.g., PFA-100) (39,40). The latter can be used to provisionally identify a platelet defect, or anti-platelet medication affect (e.g., aspirin), but comprehensive platelet testing is required to identify and characterize specific qualitative platelet disorders. This latter process is complex and technically demanding, and involves challenging patient material either as platelet rich plasma (using light transmission aggregometry) or as whole blood (using a whole blood aggregometer) with various platelet agonists (11–14,41).
Thrombophilia testing
Laboratory testing may include assays to detect congenital deficiencies or defects in the natural anticoagulants (i.e., PC, PS, AT), or acquired defects related to the presence of aPL. Congenital deficiencies in PC, PS and AT are rare, and most defects identified by laboratory testing are acquired, due to liver disease, vitamin K deficiency, DIC, or anti-coagulant medication (15). Both functional and antigenic assays are available for these test parameters, with functional assays being dominant for PC and AT (usually chromogenic assays), and antigenic assays being dominant for PS (usually LIA or ELISA). Activated protein C (APC) resistance (APCR) is defined as an impaired plasma anticoagulant response to APC added in vitro, and a relative inability to inactivate FVa and/or FVIIIa. The main molecular basis of APCR was identified in 1994 and comprises a point mutation in the gene for FV, now termed FV Leiden (FVL). There are two main laboratory methods used for testing APCR, the original APTT-based method and Russell viper venom time (RVVT) based methods (42). One of the most common and clinically important causes of acquired thrombophilia, APS, is assessed by a panel of tests that measure aPL (autoantibodies that recognize epitopes on selected proteins when bound to phospholipid surfaces). These include lupus anticoagulant (LA), which can be assessed using a variety of test processes, although the updated LA guidelines from the Scientific Standardization Subcommittee (SSC) of the International Society on Thrombosis and Haemostasis (ISTH) recommend the combined use of APTT and RVVT based assays, together with solid phase assays (43).
Factor inhibitors
Specific inhibitors can develop against any individual coagulation factor and cause a deficiency that can be detected by prolonged PT and/or APTT (44). More clinically important is that these patients can suffer serious bleeding problems (45). While some patients can develop inhibitors after treatment with plasma products (e.g., haemophilia patients that develop alloantibodies), others can develop inhibitors secondary to some other disease process (e.g., autoantibodies that lead to an acquired haemophilia), and others can be idiopathic (unknown cause). After an inhibitor is identified through mixing studies in a routine coagulation test system (i.e., PT and/or APTT, depending on which inhibitor is present or which test was prolonged), a specific inhibitor assay can be performed using a Bethesda assay or Nijmegen modification (44,46). The most common type of inhibitor is that against FVIII.
Quality control and extra-analytical issues in haemostasis testing
Unfortunately, the laboratory testing process is not problem free. There are many steps in the process of laboratory testing where the process can go awry and therefore must be controlled. In order to ensure the appropriateness of test results, laboratories are required to put several quality processes into place, and namely to undertake performance of both internal quality control (IQC) and external quality assurance (EQA). Quality control provides a process to monitor a number of variables that may include pre-analytical, analytical, post-analytical and interpretative error. For example, there are a vast number of pre-analytical variables in haemostasis. These are often beyond the control of the test laboratory, and are not always easily identifiable. As detailed below, they comprise issues that occur prior to testing and include specifics related to collection (e.g., wrong tube type, wrong collection order, sample mislabeling, etc), and transport (e.g., samples left at extremes of temperature) and inappropriate processing of the sample. Pre-analytical issues can lead to serious adverse affects on the test and thus subsequent patient care (7). Analytical variables that may cause problems in testing include inappropriate reagent/instrument calibrations, reagent issues (e.g., not properly reconstituted or used beyond stability), methodological issues (poor methodologies), and instrument problems (e.g., detector systems failing or inadequate sampling). Post-analytical and interpretative issues (7,47) relate to the interpretation of the test result by the laboratory (including the issued report) and/or the clinician (who interprets the reported test results). Incorporation of quality measures is aimed at identifying and correcting problems related to all these aspects of testing (i.e., pre-analytical, analytical and post-analytical) to ensure high quality testing results.
Preanalytical variables
Laboratory data must be accurate and reliable as erroneous results may lead to patient misdiagnosis and therefore therapeutic misadventures. Inaccuracy in laboratory reporting can arise due to problems introduced at any point in the testing process, from sample collection to report results. Due to significant advances in instruments and informatics, the analytical and reporting phases of testing do not contribute significantly to laboratory error. Rather, the pre-analytical phase of testing represents the major source of inaccurate laboratory results (48,49).
The pre-analytical phases of testing include specimen collection, transportation of whole blood specimens to the laboratory, specimen processing and storage (50). One of the most critical aspects of specimen collection is proper patient and also sample identification. Specimens must be collected in a manner to avoid in vitro clot development. Samples should be collected in a relatively atraumatic fashion and blood should flow freely into the specimen container.
Samples for haemostasis testing should be anticoagulated with sodium citrate. The World Health Organization (WHO) and CLSI recommend 3.2% sodium citrate, although 3.8% might also be acceptable. It is important that the concentration of sodium citrate is consistent within a laboratory system as clotting times tend to be longer in 3.8% versus 3.2% sodium citrate. The blood and anticoagulant must be promptly mixed to avoid in vitro clot formation. Samples for haemostasis testing that contain visible clots should not be analyzed and must be rejected. The required blood to anticoagulant ratio is 9:1 and therefore overfilling or underfilling of the evacuated tube should be avoided as this can introduce result error. Other anticoagulants such as ethylenediaminetetraacetic acid (EDTA) or heparin are not acceptable for haemostasis testing. Samples collected in EDTA or heparin anticoagulated tubes or the use of serum for haemostasis testing will lead to aberrant results and this can cause clinically significant errors in patient diagnosis. Some laboratories receive a portion or majority of test samples in secondary (aliquot) tubes and in this instance, the sample types are indistinguishable unless clearly and properly labeled as sodium citrate plasma, EDTA plasma, heparin plasma or serum. When the sample matrix is unknown or in doubt, performance of sodium, potassium and calcium assays on the sample can be undertaken to help determine the sample matrix. Samples collected in sodium citrate tend to have high sodium and low calcium values while samples collected in potassium EDTA will have elevated potassium levels with undetectable calcium values (51). Samples for haemostasis testing collected in the wrong anticoagulant tube or serum samples should not be analyzed and must be rejected.
Stability of haemostasis samples depends on which assay(s) are to be performed as well as the temperature and conditions of storage. Following collection, samples should be transported to the laboratory at room temperature promptly, ideally within one hour of drawing. Cold storage of citrated whole blood, either by placing samples in an ice bath or refrigerated (2–8 ºC) may lead to activation of platelets, activation of factor VII and significant time dependent loss of both FVIII and VWF. Depending on the tests ordered and whether unfractionated heparin is present in the specimen, samples can be stored either as whole blood or centrifuged and the plasma left remaining on the red cells or the plasma aliquoted.
In summary:
Samples for platelet function testing should remain at room temperature and testing completed within 3 to 4 hours of collection.
Samples for PT/INR evaluation are stable at room temperature for 24 hours. Samples can be stored as whole blood or centrifuged.
Samples containing unfractionated heparin must be processed within 1 hour of collection because of release of PF4 from platelets in vitro which neutralizes the heparin leading to spuriously low heparin levels as measured by an APTT or anti-FXa assay. If the whole blood is centrifuged within 1 hour, the heparinized plasma can be left at room temperature for up to four hours prior to testing.
Samples for APTT testing that do not contain unfractionated heparin and samples for special coagulation assays should be maintained at room temperature if testing will be complete within four hours. Samples that cannot be tested in this time frame should be spun and the plasma frozen in a freezer that does not undergo automatic freeze thaw cycles.
Samples should be centrifuged to obtain platelet poor plasma containing ≤ 10 x 109 platelets/L. In order to ensure appropriate removal of platelets for some particularly sensitive tests including LA, a process of double centrifugation is recommended (7,43). This is especially important if the plasma will be frozen prior to testing. The use of micropore filters to remove platelets is not recommended since this filtration process can lead to loss of factors V, VIII, IX, XII and VWF (43,54). Samples that are stored frozen should remain at –20 ºC for no more than two weeks. Prior to testing, samples should be completely thawed in a 37 ºC water bath (for about 5 minutes) and mixed thoroughly prior to testing. Repeated freeze/thaws should be minimized, to retain sample integrity.
Internal quality control (IQC)
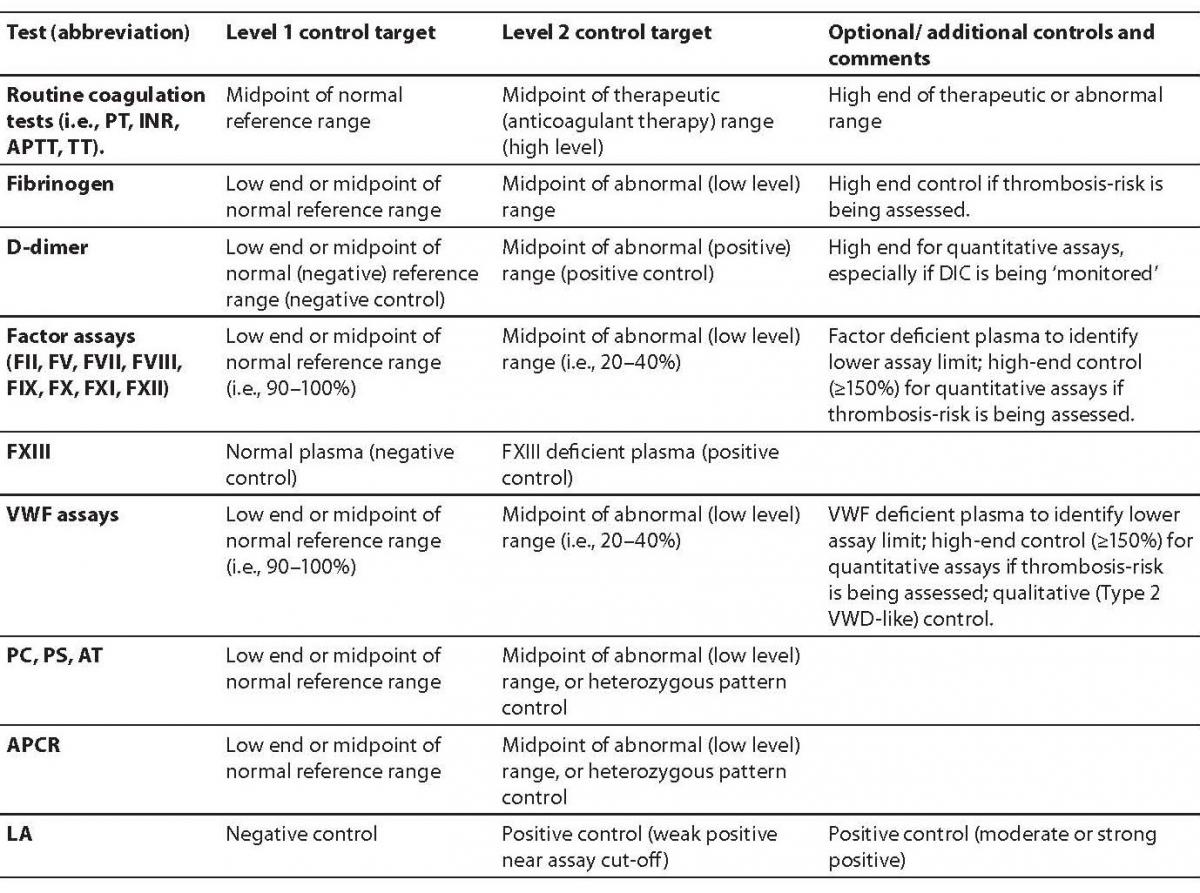
IQC must be analyzed with each assay performed within the laboratory. The purpose of IQC is to assess ongoing assay performance to ensure that assays are performing according to specifications to ensure results that are accurate and reliable. IQC material generally includes ‘normal’ and ‘abnormal’ sample controls with results plotted using a Levy-Jennings chart. This process is performed for each analyte tested (i.e., PT, APTT, etc), and using a time-frame suited to each test performed. For example, for continuous test systems such as APTT, it might be appropriate to perform testing every two, four or so hours. For tests performed in batches, it might be appropriate to perform controls at the start, middle and end of the test run. When IQC falls out of the expected range, laboratories can refer to several rules (e.g., Westgard Rules; available at:www.westgard.com) to determine if the assay is still performing adequately or if instrument/assay troubleshooting is required. For example, when IQC results are flagged as being out of the expected range by more than three standard deviations, the laboratory should be alerted and the problem investigated and resolved. Results from that particular run should not be reported until the out of range IQC issue is addressed. Records should be kept of changes in lot numbers of reagents and variation between reagent lots monitored. Recalibration may be required for specific assays (e.g., new standard curve for specific factor assays). IQC can be readily and easily applied to most diagnostic haemostasis assays. Several levels of controls are utilized for each and every test analyte. Some examples are given in Table 1.
Table 1. Some quality control specifications for coagulation and haemostasis.
In contrast, a review of the IQC (and EQA) for platelet aggregation testing emphasizes the struggle diagnostic laboratories may face providing adequate quality control for particular tests. In this instance, for example, one issue comprises the collection and transport of fresh blood to provide functional platelets for testing. An important function of IQC is to ensure reproducibility of testing. For platelet aggregation this should therefore ideally include a normal (and ideally an abnormal) control with every batch of platelet functions performed. A normal (and an abnormal) control should also be run with each new lot of reagents. Although this may seem simple it is not without its challenges. Commercial control material does not exist. Abnormal control material would need to be obtained from patients with known defects, or on anti-platelet agents. The provision of normal donors to provide fresh platelets is also an issue, particularly for small laboratories where ‘volunteers’ may be in short supply. There are also obvious ethical issues related to these invasive procedures being regularly repeated on normal and patient ‘volunteers’. The current CLSI guidelines (52) state; “It is good laboratory practice that [platelet function] reagents are tested with a normal platelet control each time an aggregation study is performed; one may plot these cumulative values on a QC chart to assess their potency”. However, while this can be achieved in large laboratories (55), it is not typically feasible for most test laboratories. Other international guidelines on platelet function testing aiming to provide diagnostic laboratories with guidance on sample collection, processing and analysis are currently being formulated by members of the ISTH.
External quality assurance
EQA describes a different and supplementary process to IQC. EQA is a peer group assessment process that allows a laboratory to assess individual analytes against those of other laboratories using either the same or different reagents or instrumentation. EQA also provides a means to evaluate long-term performance of laboratories. For haemostasis testing, the analytes assessed by EQA programs are those used in the diagnosis and treatment of bleeding and thrombotic disorders. As well as providing an external assessment of laboratory test performance, most EQA providers also offer educational support for laboratories, either through expert advice or via the distribution of written publications. In addition, most EQA providers distribute and collate questionnaires related to current practice, which also inform participants on various topics of interest, and provide additional information on reagents, instrumentation and laboratory practices.
Assessment criteria utilized by EQA providers may vary from program to program. EQA reports typically permit laboratories to also undertake self-assessment of their own results compared to those of their peers. Individual laboratory bias and drift can be monitored as well as accuracy and precision. For example, the Royal College of Pathologists of Australasia (RCPA) Haematology Quality Assurance Program (QAP) provides participants with summary data for an entire year’s program to give an overview of long-term laboratory performance. End of cycle (EOC) reports also provide a statistical analysis of the results for each analyte over the survey year.
Participation in a program that offers EQA is often a requirement for laboratory accreditation by regulatory authorities (56). For example, within Australia, medical laboratory accreditation is conducted by a body called NATA (National Association of Testing Authorities), which assesses all Australian diagnostic haemostasis laboratories on a three-year basis. In the U.S. this is largely accomplished by the College of American Pathologists (CAP). During these assessments, the results from the EQA program are examined and any analytes outside of defined ‘allowable limits of performance’ need to be identified as being actioned or investigated by the laboratory, and an explanation of any discrepancies needs to be recorded. If the problem occurs over more than one survey then the laboratory needs to be proactive in solving the issue. Moreover, the EQA providers are themselves also monitored by these government regulatory bodies. For example, the RCPA Haematology QAP is accredited by NATA and also assessed on a three-year basis.
Importance of external quality assurance
There are many internationally recognized EQA programs, although several are prominent in the area of coagulation and haemostasis (57–60). These include the RCPA Haematology QAP, the UK-based NEQAS (National External Quality Assurance Scheme), the European based ECAT, and the US based CAP (College of American Pathologists) and NASCOLA (North American Specialized Coagulation Laboratory Association). There are also several National programs that conduct EQA programs within more localized regions such as individual countries. All programs have value, although economies of scale for some analytes would favor the larger or international programs. Thus, EQA for routine coagulation testing would largely be covered by all EQA programs that offer coagulation-based programs, although local or National programs would likely dominate, whereas assessment of analytes for the more specialized tests of haemostasis (e.g., VWF, factor inhibitors, etc) would only be provided by some of the international EQA programs.
In any case, it is through these EQA programs that laboratories can monitor, improve and establish good laboratory practice in order to ensure accuracy, precision and quality of testing. This in turn will provide more correct diagnoses and safer patient care by clinicians, which is the main purpose of laboratory procedures. The following paragraphs highlight some recent initiatives by various EQA providers.
Routine coagulation testing
Most publications about the evaluation of routine coagulation testing relate to their application within the investigation of specific disorders (see following sections) or to INR monitoring of anticoagulant therapy or to D-dimer testing. With respect to the both INR and D-dimer, many ongoing issues have been highlighted by EQA providers. For example, errors in the equation used to calculate INR or errors in the conversion of units used in reporting quantitative D-dimer assays (57–62).
Thrombophilia testing
Since both test methodology and the composite tests performed within a panel can affect the accuracy of the detection of specific thrombophilia markers, assessment of testing practice by evaluation of laboratory based test proficiency using multi-laboratory surveys (EQA) is considered a basic necessity in laboratory practice.
Lupus anticoagulant (LA)
Because of the heterogeneity of LA antibodies and the dependence of the laboratory testing on the clinical diagnosis, reliability in the many different assays used to detect aPL is needed. Test performance guidelines have recently been updated by the ISTH SSC (43). Despite these efforts, testing remains plagued by a plethora of methodological issues and non-standardization in test practice (18). The CAP published an evaluation on the effect of LA on routine coagulation factor assaysin 1986 and 1987 (63). At that time, the APTT reagents varied in their sensitivity to LA leading to the conclusion that laboratories needed to be careful in their selection of APTT reagents for detecting LA. Also noted was the potential for a strong LA to interfere with the FVIII assay causing falsely low levels for all APTT reagents with a high titre LA.
Over a period of three years, NEQAS evaluated results returned by participants for a weak, strong and absent LA samples (64). The testing was performed in 220 centers and the APTT, dRVVT (dilute RVVT) and KCT (kaolin clotting time) results were evaluated. The ability of individual laboratories to make an appropriate diagnosis was influenced by selection and application of the test technique. The presence of a strong LA was not detected by 4% of laboratories, with the correct diagnosis being made by 94% of dRVVT users and 85% of KCT users. A weak LA was not detected by over half of the testing centers. Improvements in testing for LA were found by this EQA provider in a later survey after dissemination of national guidelines on laboratory methods (65).
The RCPA Haematology QAP has over the last 10 years evaluated over 20 LA survey samples distributed to participants (most were various grades of positive for LA, a few were normal or equivocal). Most laboratories used a panel of tests including the APTT (100% of participants), KCT (26–76%), and dRVVT (75–100%). Coefficients of variations (CVs) for assays have ranged from 5 to 40%. dRVVT appeared to be the most sensitive and specific for detection of LA, although laboratories performed best when they used a panel of tests (66). The overall error rate was generally < 5%, and was in part dependent on the strength of the LA (higher rates for weaker LA). Although the then-current LA guidelines were followed by the majority of participant laboratories, interpretative errors were often produced by the small number of laboratories that did not.
NASCOLA and ECAT have also assessed and reported on performance of LA testing, with NASCOLA recently identifying a high false negative rate of 35–40%, and identifying that some 25% of laboratories did not comply with ISTH recommendations(67).
AT, PC, PS and APCR
A diagnostic error rate of 2–10% has been observed for AT, PC, PS, and APCR from participants of the RCPA QAP, with both test type and test methodology contributing to this (66). The best performer is the AT assay, while the worst are the PS assays (both total and free PS assays) with CVs reaching 30%. Tests for PC also suffer from some degree of variability, and both PC and PS clot-based assays may on occasion be influenced by the presence of APCR. Similar findings have also been reported by the NEQAS and ECAT (68,69).The APCR assay error rates were greater for the APTT-based method (even when FV deficient plasma was added to the test system) than the RVVT-based method, which performed well even without the addition of FV deficient plasma (66).
Factor inhibitor investigations
This has recently been reported by various EQA providers (70–76). In brief, high inter-assay variability in assay results has been consistently observed in all reports, with little or no improvement over the past decade. Also concerning is the lack of standardization across laboratories, with no two laboratories identified as performing the same test procedure in a recent evaluation from the RCPA Haematology QAP (72,73). Overall, the Nijmegen method appears to perform slightly better than a standard Bethesda method.
Von Willebrand factor/ von Willebrand disease investigations
In the period of 2003–2005, CAP evaluated the performance of 160 to 219 laboratories using three different lyophilized plasmas with VWF antigen levels of 25%, 50% and 100% (77). The results showed high accuracy for the VWF:Ag and VWF:RCo activity assay, with all-method mean values falling between 3.2% and 5.6% respectively. However, whilst the VWF:Ag assay showed moderate precision, with all CVs ranging from 10.7% to 15.1%, the VWF:RCo assay showed low precision, with all method CVs ranging from 23.3 to 30.9%. Precision increased as the VWF concentration increased for all assays.
The RCPA Haematology QAP has dispatched a total of nearly 60 plasma samples to participants for VWD evaluation over the years 1998 to 2009 (34,35,78). These samples have included plasma from normal individuals plus all of the major VWD subtypes (Type 1, Type 2A, 2B, 2M, 2N, and Type 3). FVIII:C was performed by all participating laboratories. VWF:Ag was performed by all participants in earlier surveys, but in more recent survey a few laboratories have not included this in their panel of tests. The VWF:RCo assay was performed by > 90% of participants in earlier surveys, but this has dropped to ~ 60% of participants in more recent surveys, indicating that many laboratories have ceased using this assay. Usage of the VWF:CB assay has been fairly consistent over the years with ~ 50% of participants performing the assay. Greater than 80% of participants generally perform at least one functional assay (i.e., either VWF:RCo or VWF:CB) and in ~ 40% of cases perform both assays. The CVs for all these assay has been reported in the literature and are dependent on both the methodology and level of VWF in the sample, but tend to increase in the order FVIII:C < VWF:Ag < VWF:CB < VWF:RCo, and CVs are highest when the VWF level is lowest. In the more recent reports, the issue of diagnostic interpretive error rates associated with methodology and test panels has been explored. The main finding is that addition of VWF:CB to a core test panel of FVIII:C, VWF:Ag, and VWF:RCo will consistently and substantially reduce VWD diagnostic error rates.
Another major recent issue explored by the RCPA Haematology QAP was the evaluation of the performance of the VWF tests at (diagnostically important) low levels of VWF (35). The RCPA QAP identified the lower limit of sensitivity for VWF:Ag and VWF:CB assays to be up to 10 U/dL, but this was up to 20 U/dL or greater for VWF:RCo. Interestingly, automation of many of these test procedures over the last few years has not improved the lower limit of sensitivity performance in the diagnostic haemostasis laboratory. The RCPA Haematology QAP made several recommendations to help reduce the possibility of diagnostic error in VWD related to this issue, and namely:
1. choose appropriate methodologies capable of measuring levels of VWF well below 20 U/dL;
2. repeat VWF testing on a fresh sample for confirmation;
3. use a comprehensive range of appropriate controls;
4. adjust the concentration of the test plasma to match assay tolerances; and
5. perform as comprehensive a test panel as possible.
Other EQA providers have reported on VWF testing, and have also shown considerable variation in test practice and in test results, both of which compromise the diagnostic process (79,80).
Platelet function testing
Although not ideal, recent initiatives from the RCPA and other EQA providers show that some form of EQA for platelet function testing is also feasible (81–87). As an example, the RCPA is offering a new program to cater to the PFA-100 analysis in laboratories from 2010.
Guidelines for improving the quality of coagulation and haemostasis testing
A number of organizations provide guidelines to improve the testing process for coagulation and haemostasis. These include various ISTH SSCs, and CLSI. Thus, ISTH SSC guidelines for LA testing have recently been published (43), as have some guidelines related to platelet function testing using the PFA-100 (88). The CLSI is also very active in this arena, and has published a wide variety of relevant guidelines, including those for collection and processing of coagulation samples (50), for generating normal reference ranges in testing (89), as well as for platelet function (52), VWF and factor testing (90; currently under revision), and PT and APTT testing (91). Other guidelines are in progress, including one on the Quantitative D-dimer for Exclusion of Venous Thromboembolism (currently in draft form) and one on LA testing.
Conclusion
The haemostasis pathway is very complex, and includes various elements of activation and regulation. The investigation of aberrant haemostasis or the monitoring of anticoagulant therapy requires the performance of a multitude of test procedures. These in turn can be influenced by many factors ranging from anticoagulant therapy, liver disease, and infections, in addition to congenital and acquired defects. Specific procedures for test collection, sample preparation, test performance, and quality assurance need to be established in every laboratory to ensure provision of accurate and reliable results to enable appropriate patient management and care. IQC and EQA facilitate this process to ensure the quality of test results. This is evidenced by various examples within this review.