Introduction
Clinical laboratories routinely use commercial diagnostic products during the testing process. Diagnostic products can be divided into two major categories: in vitro diagnostic (IVD) devices, such as laboratory instruments, reagents, assays and blood collection tubes, and medical devices, such as specimen collection devices (needles and sets). According to the United States (US) Code of Federal Regulations (CFR), IVD devices are “those reagents, instruments, and systems intended for use in diagnosis of disease or other conditions, including a determination of the state of health, in order to cure, mitigate, treat, or prevent disease or its sequelae. Such products are intended for use in the collection, preparation, and examination of specimens taken from the human body” (1). The main difference between medical and IVD devices is that IVD devices are usedin vitro for testing of samples derived from the human body, while medical devices are used in vivo.
The quality of diagnostic products is an essential element of total laboratory testing quality. Laboratory professionals expect commercial companies to manufacture high quality products. In order to meet these customer expectations, diagnostic companies have processes in place to assure that they successfully manage products throughout their life cycle, i.e.:
a) design products that meet customer needs;
b) manufacture them within developed specifications and very little variability;
c) assure safety and efficacy of their use;
d) conform with regional regulatory requirements for their release for clinical use while meeting established professional
standards and guidelines; and
e) monitor the product performance after it is released into the marketplace until its discontinuation from the market (Figure
1).
Each of these phases contains many steps, and it is possible for errors to occur at any of the numerous hand-offs, which could be detrimental for the quality of the product. In order to avoid this, diagnostic companies have developed robust operating processes supported by standard operating procedures, which are designed to eliminate variability, errors and rework.
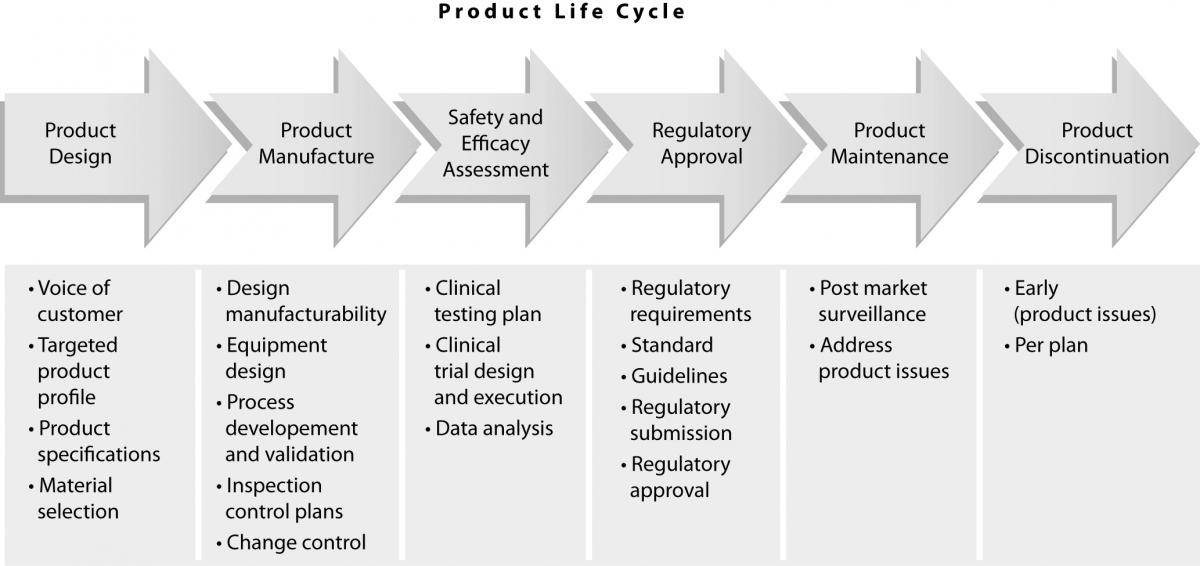
Figure 1. Product life cycle.
Product design
All diagnostic products are developed through a series of phases and gates which begin with a business idea and then develop into a new product (2). In order to design a high quality product, it is essential to address customer needs. This is usuallyaccomplished through extensive customer interactions, either directly (face-to-face interviews) or indirectly (surveys). This “voice of customer” is vital for the development product attributes, some of which are necessary to achieve expected product quality. Once this targeted product profile is defined, the product development process can proceed towards defining product specifications (tolerances), which are also an essential component of quality. If these specifications are too wide, the variability of manufactured product might exceed those needed to assure expected quality. On the other hand, specifications that are too narrow may present a problem for the manufacturability of the product, since this might be beyond the capability of the manufacturing process. During this period, it is critical to test these specifications in the context of product performance and assure that at their extremes the performance of the product is still acceptable (i.e. that the quality of the product is uniform). This is usually an iterative process, which is completed using prototypes or research reagents.
As part of this process, it is also important to select the proper materials that will be used to manufacture the product, since product quality is directly dependent on the quality of the raw materials used for its production. Once the specifications are defined, the product design is developed and manufacturability is assessed.
Product manufacturing
As mentioned previously, manufacturing quality of the diagnostic products is dependent on the consistency and repeatability of the manufacturing processes. In order to maintain this consistency, there are several key areas of focus, such as:
- Manufacturability of the Design
- Manufacturing Equipment Design
- Manufacturing Process Development and Validation
- Inspection Control Plans
- Change Control
The manufacturability of the design is an important element that will subsequently determine the complexity, capability (Cpk) and consistency of the manufacturing equipment. Many companies employ Design for Manufacturability and Assembly (DFMA) techniques to ensure the Voice of Manufacturing is heard early in the design phase. DFMA is a systematic methodology used to assess if the design can be manufactured and assembled at the targeted cost, quality and operational equipment effectiveness targets. A manufacturing friendly design will reduce the number of product design changes and manufacturing design changes that may be required post product release. Necessary design changes identified early in development are significantly less costly and time consuming than those identified in the later stages of the process. The complexity of the design (i.e. product tolerances, number of components, surface treatments) has a direct correlation on the quality of the product.
The manufacturing equipment design also has a direct impact on product quality. Equipment designs that account for the full variability in components for in the handling and assembly process result in reduced final product variability. Robust assembly testing using components at the minimum and maximum of their tolerance will ensure the component variability is factored into the equipment design.
During manufacturing process development and validation activities, the manufacturing process steps that impact the critical to quality (CTQs) characteristics of the product are defined through Designed Experiments (DOEs). DOEs are used to define the ranges of the Key Process Input Variables (KPIVs) and their impact on the CTQs. KPIVs are manufacturing process variables (pressure, temperature etc.) that have the greatest effect on output variables (impact strength, peel force) of the product being manufactured. The process validation will challenge these ranges in the operation qualification (OQ) phase to ensure the entire operating range as well as the interactions between processing parameters. Once verified through the OQ phase, the performance qualification (PQ) phase will challenge the short term capability (Cpk) of the process as well as the projected long term capability (Ppk) through multiple process runs by trained operators.
Through risk management techniques, such as Process Failure Modes Effects Analysis (pFMEA), potential process risks are identified and mitigated. pFMEA is a systematic method of evaluating a manufacturing process in order to identify potential failure modes inherent to the process, assessing the potential impact of those failure modes, and determining methods to eliminate/prevent or mitigate their occurrence. Mitigation can be accomplished through equipment design along with the inspection control plan. A properly powered inspection plan ensures that identified product CTQs are challenged at a set frequency. The inspection process must be qualified through an appropriate Measurement System Analysis (MSA) that challenges the operator influence, measuring technique as well as the variability in the parts that are measured. MSA is a specially designed experiment that seeks to identify the components of variation in the measurement.
Finally, a robust change control process for properly qualified product and process changes should be implemented. Changes to product design / materials must also be revalidated so product performance is not adversely affected. Additionally, changes to the manufacturing process need to be revalidated to ensure that validated processing parameters are unaffected as well as their impact on CTQs.
Establishment of safety and efficacy of use
Once the manufacturability of a product has been achieved, several lots of product are made using the established manufacturing process. This assesses the performance of the product in real use and is achieved by evaluating safety and efficacy (i.e. determine that the product will not cause harm to humans and will perform as intended). At the onset of this activity, it is necessary to develop a clinical testing plan which will define the scope of testing (analytes, instruments, specimen collection tubes) and is executed through investigations with human subjects or materials derived from human subjects (i.e. clinical trials). The quality of the data obtained from these clinical trials is an important component of the quality of a diagnostic product. If the clinical trials are not conducted in a controlled and regulated manner using good clinical practices, the information on product performance will not reflect its true performance in a clinical environment. First, the trial sample size, such as the number of subjects or specimens that are tested in these trials, must be sufficient (i.e. have the appropriate statistical power) to allow generation of conclusions with a high degree of certainty. Second, the trial has to be designed to generate meaningful data, and this design has to be executed tin such a manner to assure data integrity. This involves study monitoring and appropriate data management. Lastly, the data must be analyzed utilizing the statistical methodology to generate appropriate results. During this entire process, it is also imperative to guarantee the safety of the subjects that are participating in the clinical trials. Institutional Review Boards or Ethics Committees review each study design prior to implementation to assure that subjects will not be exposed to undue risk by their participation in the study. Subjects are also made aware of study risks prior to participation, since risks are disclosed in the informed consent that is signed by the participants before the start of the trial.
Regulatory requirements
Before clinical use, diagnostic products must receive approval for use from the relevant regulatory body. The approval process is highly dependent on the type of device in question. Medical and IVD devices are usually divided into different classes, based on the risk in use. It is necessary to apply the correct regulations for each device class, which vary geographically (Table 1). Typically, Class III medical devices represent the highest risk, and Class I the lowest. Depending on the device risk there are different regulatory requirements that the device manufacturers need to comply with in order to obtain approval for their distribution to the market (see Table 1). As an example, blood collection devices (needles and blood collection sets), which are medical devices most commonly used in laboratory testing, are classified as Class II (United States) and Class IIa (European Union). In the US, in order to sell a new blood collection tube, the manufacturer needs to conduct clinical trials and mechanical testing to assure that the device is safe for use. These data are submitted to the Food and Drug Administration (FDA) for approval prior to launch. According to the EU Directive, it is enough that the manufacturer has the product performance and assembly data on file and certification that confirms the device sterility.
Table 1. Classification of medical devices.

The regulatory landscape for medical and IVD devices is highly variable and constantly changing. In some regions, regulatory requirements are evolving and transparent, and in others, static and unclear. Most major markets are strictly regulated with an emphasis on pre-market control.
In today’s rapid business environment, it is important to utilize standards for assuring product compliance and to facilitate acceptance of medical devices worldwide.
This is also increasing the demand for better regulatory guidance to ensure that products are safe and effective before entering the market.
FDA relies on the current Good Manufacturing Practice (GMP) requirements of 21 CFR part 820 Quality System Regulation (QSR). Outside the medical industry, the international standards of the Quality Management System (QMS) are based on ISO 9001, and for the Medical Device industry, ISO 13485. ISO 13485 “Medical Devices, Requirements for Regulatory Purposes” is the standard recognized by many countries as the way to obtain GMP.
The ISO 13485 standard is utilized as the QMS standard that is applicable to many regions around the world for regulatory compliance. The implementation of a QMS is required for marketing of medical devices in different regions. While the United States relies on FDA investigators to assure compliance to the QSR, other regions must rely on ISO 13485 certification for regulatory compliance.
Other more important standards for medical devices provide guidance for risk management (ISO 14971), product sterility (ISO 11607, ISO 11135 and ISO 11137), material biocompatibility (ISO 10993) and clinical trials (ISO 14155). Although ISO standards are very commonly used worldwide, there are other standards that are used by regulatory agencies to assess performance of diagnostic products. Examples are guidelines produces by the Clinical Standards Laboratory Institute (CLSI) such as EP07-A2 (3), EP09-A2 (4), EP17-A (5), EP25-A (6), GP34-P (7). For instance, EP07-A2 outlines procedures for manufacturers to screen potentially interfering substances, quantify interference effects, and confirm interference in patient samples. On the other hand, EP17-A provides guidance for determining the lower limit of detection of clinical laboratory methods, for verifying claimed limits, and for the proper use and interpretation of these limits. Many countries outside of Europe and the United States depend on regulatory standards to assure their organization’s compliance, as they do not have adequate resources to conduct inspections outside their borders.
The ISO 13485 certification assures that medical device companies comply with a known regulatory standard. The QMS of the medical device company is also certified by a registrar who routinely audits the organization to verify that compliance is maintained.
Maintaining the ISO certification is only a part of the process. Regionally, there are various requirements and additional processes must be in place. Some of these include regulatory submission activities, adverse event reporting and communications with regulatory agencies.
A variety of International Standards are utilized by many companies to address business, quality, clinical and regulatory needs, and play a significant role in raising the levels of safety, reliability and efficiency. International standards are also useful to industrial and business organizations, governments and regulatory bodies and ultimately, the end user. These standards provide an implicit pathway of ensuring a level of product quality.
Post market surveillance
Once the product is launched, it is important to monitor its performance over time in order to ensure constant product quality. This is known as post market surveillance. It can be active, such as conducting occasional studies to assess performance or by solicitating customer feedback or passive by monitoring customer complaints (Figure 2). Regardless of the way that the feedback on product performance is obtained, if the indication of product malfunction is confirmed, IVD companies immediately assess the degree of risk that this malfunction can pose on the safety and efficacy of the device as well as its root cause. If this risk is deemed unacceptable, the product is withdrawn from the market and steps are taken to assure that this does not happen again. Product malfunction is usually due to a manufacturing error, in which case processes are put in place to prevent this error. However, it can sometimes result from an inherent flaw of the device. In that case, redesign might be the only solution.
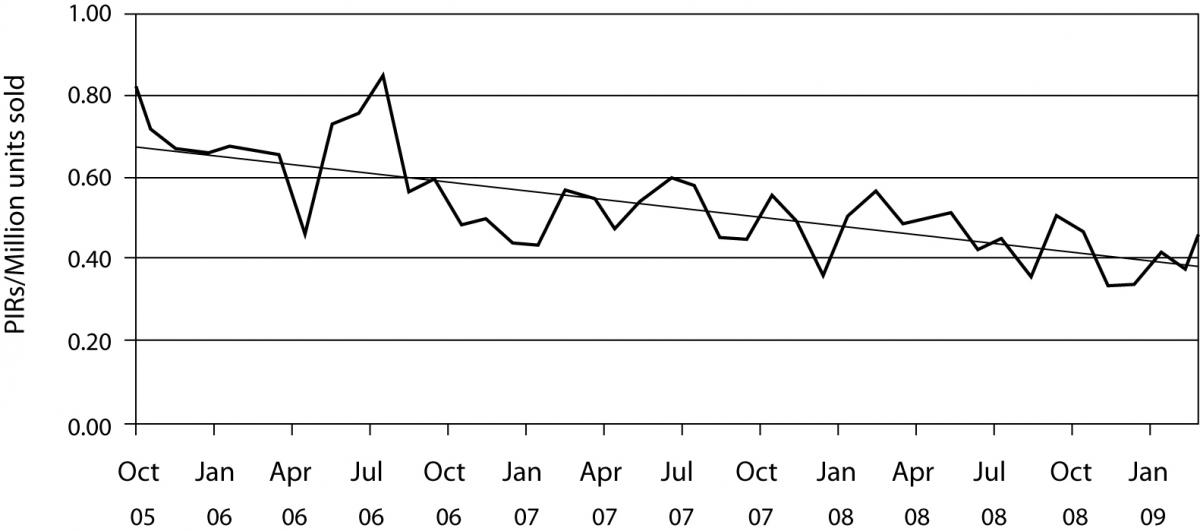
Figure 2. Product incident report (PIR) rates. Worldwide complaint rates for all products for BD Diagnostics – Preanalytical Systems. Presented are monthly complaint rates and the linear complaint rate in PIRs / million units sold.
So far, we have described processes that IVD companies use to assure quality of their offerings. However, often this is not sufficient to achieve total quality in laboratory diagnostics. Owing to technology advancements, analytical instruments for the clinical laboratory are becoming increasingly sophisticated and new and more sensitive assays and reagent tests are developed ever faster. Therefore, quality systems for monitoring laboratory test accuracy and efficiency need to focus beyond just the performance of individual analytical devices to encompass the entire laboratory testing process. Laboratory professionals should obtain accurate results from “the system”, including sample collection tubes, instruments and reagents. The highest level of quality is achieved only when instrument/assay manufacturers and sample collection manufacturers cooperate to deliver total system performance. Because of this, there is a rising need for the manufacturers of the instruments, reagents/assays and the sample collection devices (such as evacuated tubes and sharps) to work together to deliver total system performance (Figure 3).
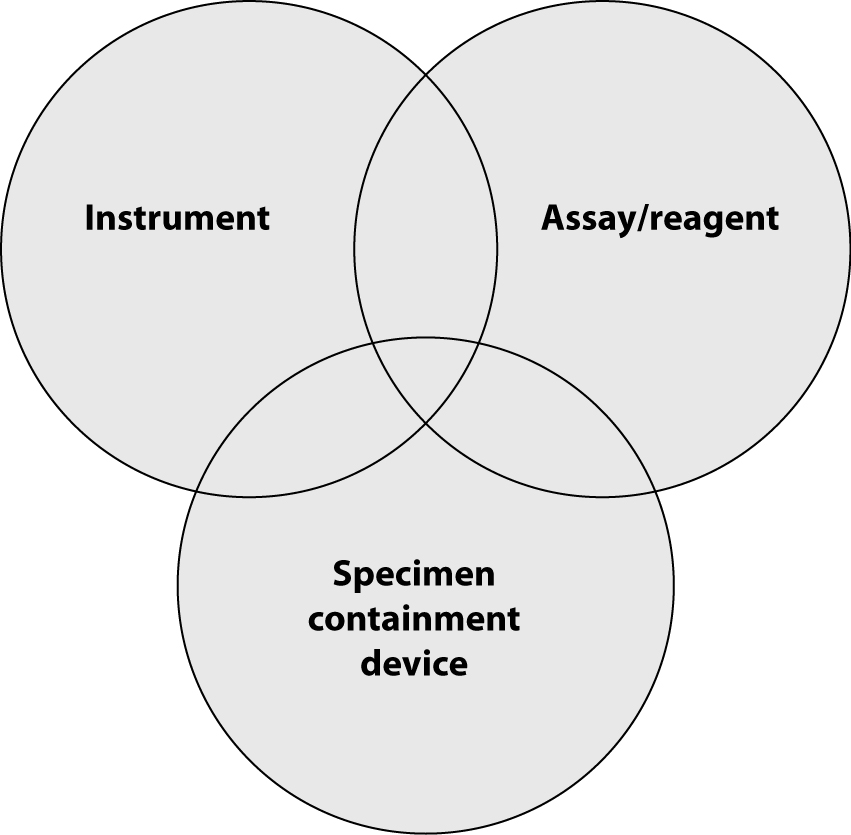
Figure 3. Total system performance.
Device manufacturers, tube vendors, and clinical laboratory professionals alike need to understand the interactions between tubes, reagents and instruments and the contribution of each to total system performance and subsequently, accurate results. Analytical device manufacturers and specimen containment vendors must work as partners during installation of new laboratory systems and validate the entire laboratory testing process from sample collection to reporting of results.
It may not be always practical or even possible for tube manufacturers to test their products on all assay platforms. Therefore, consistency of tube additives and the effect of all tube components on assay performance and test results throughout the dynamic range of the assays should be verified on at least two different instrument platforms. An array of analytes for testing should be selected using several criteria:
a) medical significance;
b) frequency of use;
c) assay methodology;
d) chemical structure, etc.
in order to identify potential interferences. Similar strategy should be applied to the selection of instrument platforms for testing. Likewise, when a new assay or instrument platform is under development, the manufacturer should evaluate the performance with specimens collected in tubes from the major global suppliers of sample collection devices and assess incompatibilities that occur inside and outside the tube. Additive components present inside the tube can affect assay performance. On the other hand, the mechanical components of the specimen container, such as diameter, height, wall material, stopper configuration and material have to be compatible with the mechanical components of the instrument analyzers. Additionally, the manufacturers of specimen containment devices should evaluate new tubes under stressed conditions – partially filled tubes, extreme temperatures and shipping conditions, and prolonged contact time of the sample with the tube additives – to simulate “real life” conditions.
Before launching a new specimen collection device to the market, specimen containment manufacturers should work together with instrument and/or assay manufacturers to achieve total system performance, optimum efficiency and TAT. IVDcompanies that are concerned about total quality in laboratory diagnostics have dedicated resources to assure total system performance. These individuals collaborate with colleagues from instrument/assay/tube companies to address particular customer complaints and find root cause and ways to mitigate it. They are also involved in sharing information about new product generation maps and working with their respective Research and Development groups to assure mutual compatibility of products. In addition, total system performance should be demonstrated based on joint clinical studies and shared with customers. When issues arise, such as assay interferences, it is crucial that all vendors who constitute a particular laboratory system work with their joint customers to resolve the issue. Total quality in laboratory diagnostics can only be achieved through monitoring of total system performance.
As described, IVD companies have instituted distinctive processes that are designed to assure maximum quality of their offerings. Many of these processes are overlapping and contain multiple checks and balances that were designed to prevent erosion of safety and product quality. This built-in redundancy is deliberate, as it is a way to assure steady maintenance of product quality through multiple hand-offs during the product life cycle. On the other hand, regulators play an important role in maintaining needed IVD product quality by setting performance standards enforced through formal product approval processes before allowing their distribution in the marketplace. As discussed previously, additional focus is now centered on total system performance which includes the interaction of the laboratory instrument, the assay and the specimen containment device.
Acknowledgements
The authors would like to thank Julie Ravo for her work on editing this manuscript.