Introduction
Inflammation and haemostasis are tightly interrelated pathophysiologic processes that considerably affect each other. In this bidirectional relationship, inflammation leads to activation of the haemostatic system that in turn also considerably influences inflammatory activity (1,2). Close interaction between immune and haemostatic systems is an attempt to restore normal tissue function following injury. Local activation of the haemostatic system is an essential part of the host defence in both infectious and non-infectious inflammatory states. However, an exaggerated and insufficiently controlled haemostatic activity induced by inflammation may appreciably contribute to disease severity. The two examples of clinical conditions in which the tight interdependent relationship between inflammation and haemostasis considerably contribute to the pathogenesis and/or progression of disease are systemic inflammatory response to infection or sepsis and acute arterial thrombosis as a consequence of ruptured atherosclerotic plaque. Close links between inflammation and haemostasis help explain the prothrombotic tendency in these two clinical conditions. Inflammation shifts the haemostatic activity towards procoagulant state by the ability of proinflammatory mediators to activate coagulation system and to inhibit anticoagulant and fibrinolytic activities. Once the activation of haemostatic system occurs in inflammatory states, amplification of the haemostatic disorder can result in thrombosis and organ damage. In turn, uncontrolled activation of the haemostatic system can also amplify the initial inflammatory response thus causing additional organ injury. Such, the haemostatic system acts in concert with the inflammatory cascade creating an inflammation-haemostasis cycle in which each activated process promotes the other and the two systems function in a positive feedback loop.
The goal of inflammation is to restore the integrity of threatened or damaged tissues because of injury or infectious pathogens. On the other hand, haemostasis is a physiological defence mechanism to stop bleeding due to vessel wall damage (3). Activated by vessel wall injury, the haemostatic system consists of a complex network of individual components. Vascular endothelial cells (ECs) and platelets constitute primary haemostasis resulting with a formation of platelet clot. This process is followed by an activation of the plasma coagulation cascade, so called secondary haemostasis that induces a formation of a fibrin clot. The net result of the haemostatic activation is stable platelet-fibrin clot. These processes are firmly regulated by physiologic anticoagulant mechanisms that keep activation of the coagulation system under control and by fibrinolytic system which is responsible for degradation of the platelet-fibrin clot which has fulfilled its function (4).
In recent years, various molecular mechanisms that play a role in the bidirectional relationship between inflammation and haemostasis have been intensively investigated and elucidated. This review summarizes the current knowledge of the complex interactions in the bidirectional relationship between these two interrelated processes.
Mechanisms by which inflammation induces disturbance of the haemostatic system
The extensive crosstalk between immune and haemostatic systems occurs at the level of all components of the haemostatic system including vascular ECs, platelets, plasma coagulation cascade, physiologic anticoagulant pathways and fibrinolytic activity. During an inflammatory response, inflammatory mediators, in particular proinflammatory cytokines, play a central role in the effects on haemostatic system (5,6). The main mediators of inflammation-induced activation of the haemostatic system are proinflammatory cytokines tumour necrosis factor-alpha (TNF-a), interleukin 1 (IL-1) and interleukin 6 (IL-6) (7). Inflammatory mediators trigger disturbance of the haemostatic system in a number of mechanisms including endothelial cell dysfunction, increased platelet activation, tissue factor (TF) mediated activation of the plasma coagulation cascade, impaired function of physiologic anticoagulant pathways and suppressed fibrinolytic activity (Figure 1).

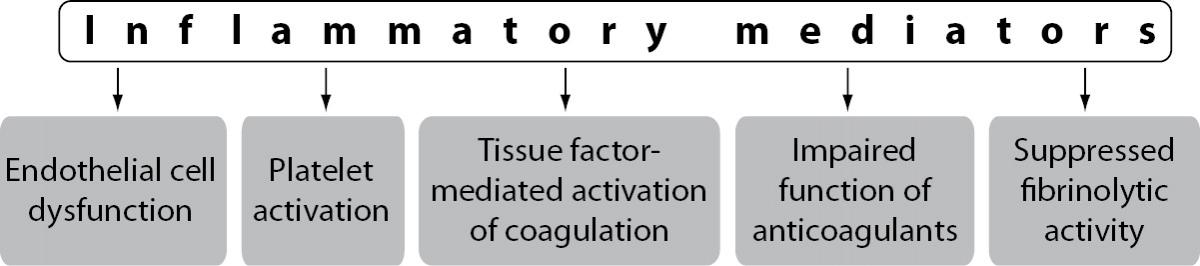
Figure 1. Mechanisms of inflammation-induced disturbance of the haemostatic system.
Dysfunction of vascular endothelial cells
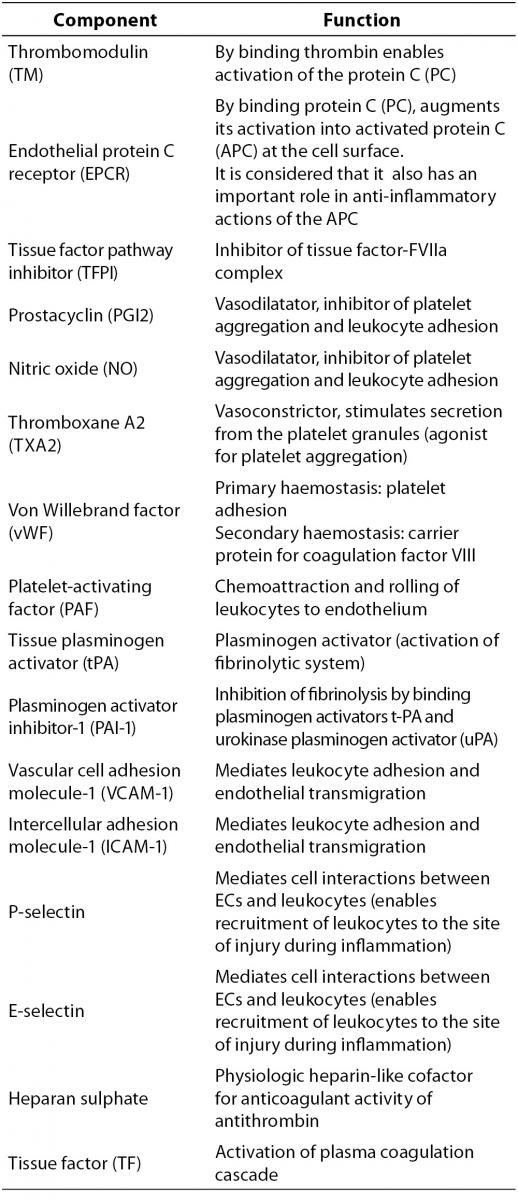
Vascular ECs are actively involved in haemostasis. In physiological conditions, intact endothelium functions as an antithrombotic surface thus preventing inappropriate activation of coagulation. ECs are the main producer of components with proinflammatory, procoagulant and antifibrinolytic activities, as well as of those with the opposite actions, i.e. anticoagulant, anti-inflammatory and profibrinolytic properties (Table 1). All these components are essential in maintaining a physiological balance involved in the regulation of the haemostatic system.
Table 1. Integral components of the vascular endothelial cells.

Irrespective of its aetiology, inflammation leads to an imbalance between proinflammatory and procoagulant and anti-inflammatory and anticoagulant properties of the endothelium thus contributing to disturbance of the haemostatic system. Once activated or injured, ECs secrete into the local environment mostly procoagulant or antifibrinolytic components, such as von Willebrand factor (vWF), thromboxane A2 (TXA2) or plasminogen activator inhibitor-1 (PAI-1)) while the expression or secretion of components with anticoagulant and profibrinolytic properties is significantly reduced (Figure 2). In addition, activated ECs can express at their surface tissue factor (TF) and adhesion molecules such as P- and E-selectins, vascular cell adhesion molecule-1 (VCAM-1) and intercellular cell adhesion molecule-1 (ICAM-1) (Figure 2). Adhesion molecules have important role in mediating interaction of neutrophils and platelets with endothelium, thus also promoting the inflammatory and haemostatic responses. As a result of disturbed balance due to inflammation, the function of ECs shifts from an anticoagulant, anti-inflammatory and vasodilatatory phenotype to a proinflammatory and procoagulant state. Such, in inflammatory states the endothelium serves as an important interface between inflammation and inappropriate activation of the haemostatic system (8). Proinflammatory cytokines TNF-a, IL-1 and IL-6 are crucial in mediating the procoagulant effects of dysfunctional ECs. Vascular ECs respond to the cytokines expressed and released by inflammatory cells, such as activated leukocytes, but can also release cytokines themselves thereby amplifying the haemostatic response in inflammatory states (9).

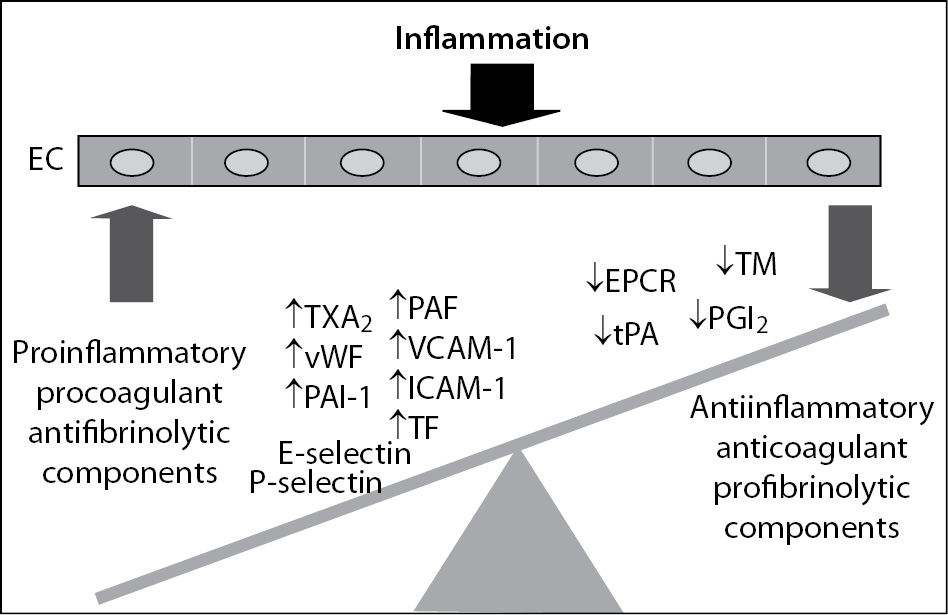
Figure 2. Endothelial cell dysfunction induced by inflammation. EC - endothelial cell; TXA2 – thromboxane A2; PAF – platelet-activating factor; vWF – von Willebrand factor; PAI-1 – plasminogen activator inhibitor-1; VCAM-1 – vascular cell adhesion molecule-1; ICAM-1 – intercellular adhesion molecule-1; TF – tissue factor; EPCR – endothelial protein C receptor; TM – thrombomodulin; tPA – tissue plasminogen activator; PGI2 – prostacyclin.
ECs play an important role in all mechanisms that contribute to inflammation-induced disturbance of the haemostatic system, including activation of platelets and other inflammatory cells (in particular leukocytes), TF-mediated initiation and regulation of thrombin generation, as well as impaired function of physiologic anticoagulants and suppressed fibrinolytic activity. Physiologic inhibitors of coagulation, such as protein C (PC) system and tissue factor pathway inhibitor (TFPI) are connected to the endothelium, so that ECs play a key role in maintaining the function of these anticoagulant mechanisms. Also, as the main producer of fibrinolytic activator, i.e. tissue plasminogen activator (tPA) and inhibitor (PAI-1), ECs also have an obvious role in suppressed fibrinolytic activity in inflammatory states. All these changes of ECs lead to the exposure of procoagulant components to blood and contribute to a prothrombotic state in inflammatory states.
Platelet activation induced by inflammation
Beside of an important role in haemostasis, platelets also play a relevant function in inflammation acting as proinflammatory cells (10). In physiological conditions, platelets circulate in a resting state, protected from activation by inhibitory mediators, such as nitric oxide (NO) and prostacyclin (PGI2) released from intact ECs. Numerous factors promote platelet activation during inflammatory response. Such, EC dysfunction leads to increased platelet reactivity due to its elevated production of TXA2 and vWF and decreased production of PGI2. Upon vessel wall injury platelets rapidly adhere to the exposed subendothelial matrix. This process is mediated by several cellular receptors as well as various adhesive and regulatory proteins present on platelets or ECs thus allowing platelet-vessel wall interaction and subsequent thrombus formation (11). Additionally, in the setting of inflammation-induced activation of haemostasis, platelets can be directly activated with inflammatory mediators such as proinflammatory cytokines or platelet-activating factor (PAF) in case of both non-infectious and infectious inflammatory states (12) or with endotoxin in case of sepsis. Further, thrombin generated by activated coagulation cascade is one of the strongest platelet activators (Figure 3).

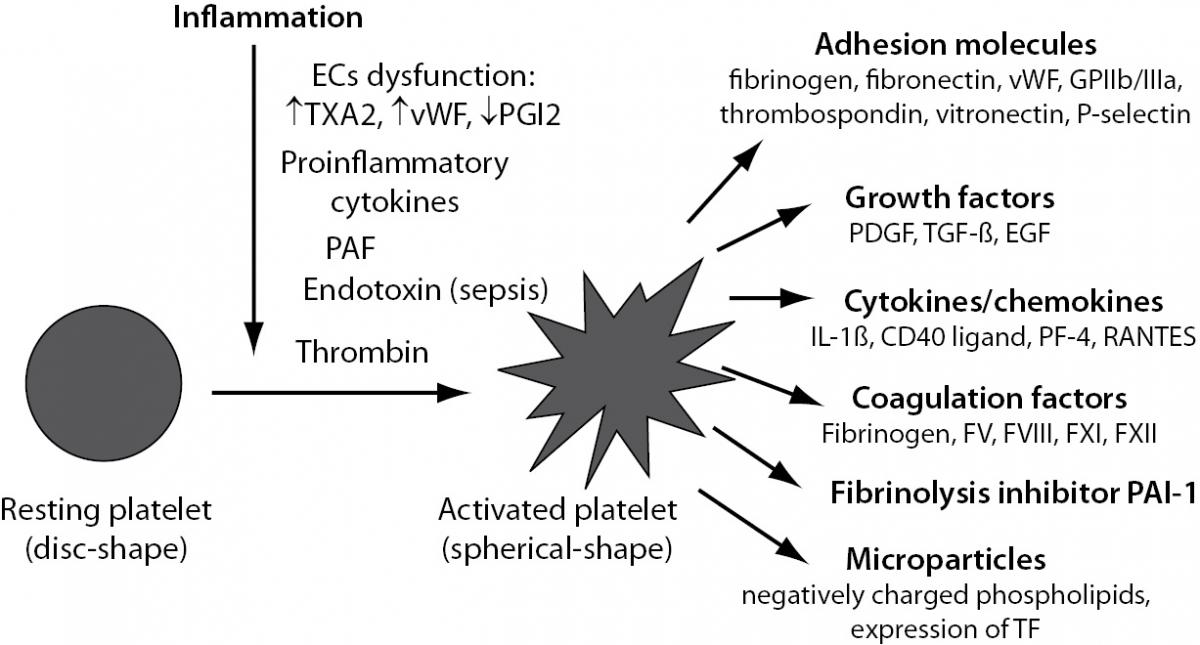
Figure 3. Platelet activation induced by inflammation. EC – endothelial cell; TXA2 – thromboxane A2; vWF – von Willebrand factor; PGI2 – prostacyclin; PAF – platelet activating factor; GPIIb/IIIa – glycoprotein complex IIb/IIIa; PDGF – platelet derived growth factor; TGF-b - transforming growth factor-beta; EGF – epidermal growth factor; IL-1b - interleukin 1beta; PF-4 – platelet factor 4; RANTES – CCL5 chemokine; PAI-1 – plasminogen activator inhibitor 1; TF – tissue factor.
Upon activation, platelets undergo shape change from a disc-shaped cell into a spherical-shaped cell and express at their surface or secrete into the local environment a number of proinflammatory and procoagulant substances (13), such as adhesion molecules, growth factors, cytokines and chemokines and certain coagulation factors and fibrinolytic inhibitor PAI-1 (Figure 3). Additionally, platelets may generate procoagulant rich particles, called microparticles (MPs), a circulating cell fragments derived from activated or apoptotic cells. MPs contain negatively charged phospholipids essential for coagulation reactions and express TF on their surface (14,15) and thus contribute to the prothrombotic state (Figure 3).
Individual platelet mediators modulate functions of other platelets, leukocytes and ECs. As an example, activated platelets secrete IL-1β, which is a major activator of ECs. This interaction of platelets with ECs results with secretion of IL-6 and monocyte chemotactic protein 1 (MCP-1) from ECs, as well as with the expression of adhesion molecules E-selectin, VCAM-1 and ICAM-1 on ECs (16). Further, interaction of platelets with neutrophils promotes the recruitment of neutrophils into inflammatory tissue thus directly participating in the host defence (17). Interaction of platelets with inflammatory cells such as leukocytes and ECs is mainly mediated through platelet P-selectin, a cell adhesion receptor localized in platelet’s alpha-granules (18,19). Upon platelet activation, P-selectin is rapidly translocated to the cell surface and serves as the receptor for its natural ligand, P-selectin glycoprotein ligand (PSGL-1) expressed on neutrophils, monocytes and ECs thus mediating platelet-leukocyte and platelet-EC interactions (18,20). Cellular interaction via P-selectin markedly enhances the production of proinflammatory cytokines and chemokines in neutrophils and mononuclear cells and increases the expression of adhesion molecules and TF on both leukocytes and ECs (5,19). The molecular mechanism of this effect relies on nuclear factor kappa B (NF-κB) activation induced by binding of activated platelets to neutrophils, mononuclear cells and ECs (13).
Activation of the plasma coagulation cascade in inflammation
The main mechanism of plasma coagulation cascade activation in inflammation is mediated by TF (21-23). TF is a transmembrane protein constitutively expressed by a variety of cell types including circulatory blood cells and ECs. Also, it is present in all blood-tissue barriers, such as adventitial layer of the vessel wall, so that coagulation can be initiated quickly once the endothelial barrier has been disrupted (24,25). However, in contrast to other coagulation factors, TF is normally not in direct contact with blood. Such, circulatory blood cells and ECs do not express TF under normal physiological conditions. However, when the integrity of the vessel wall is disrupted or if circulatory blood cells or ECs start to express TF in response to various proinflammatory stimuli, such as proinflammatory cytokines (TNF-a, IL-1 and IL-6) or endotoxin, TF expression is induced in different cell types including leukocytes (predominantly monocytes and macrophages) and ECs. It enables that TF comes into direct contact with blood (Figure 4). On exposure to blood, TF binds to FVII and the complex TF-FVIIa initiates the activation of the coagulation cascade through the conversion of coagulation factors FIX and FX into activated forms (FIXa and FXa) thereby generating thrombin (FIIa) whose key function is to convert fibrinogen to fibrin (Figure 4).

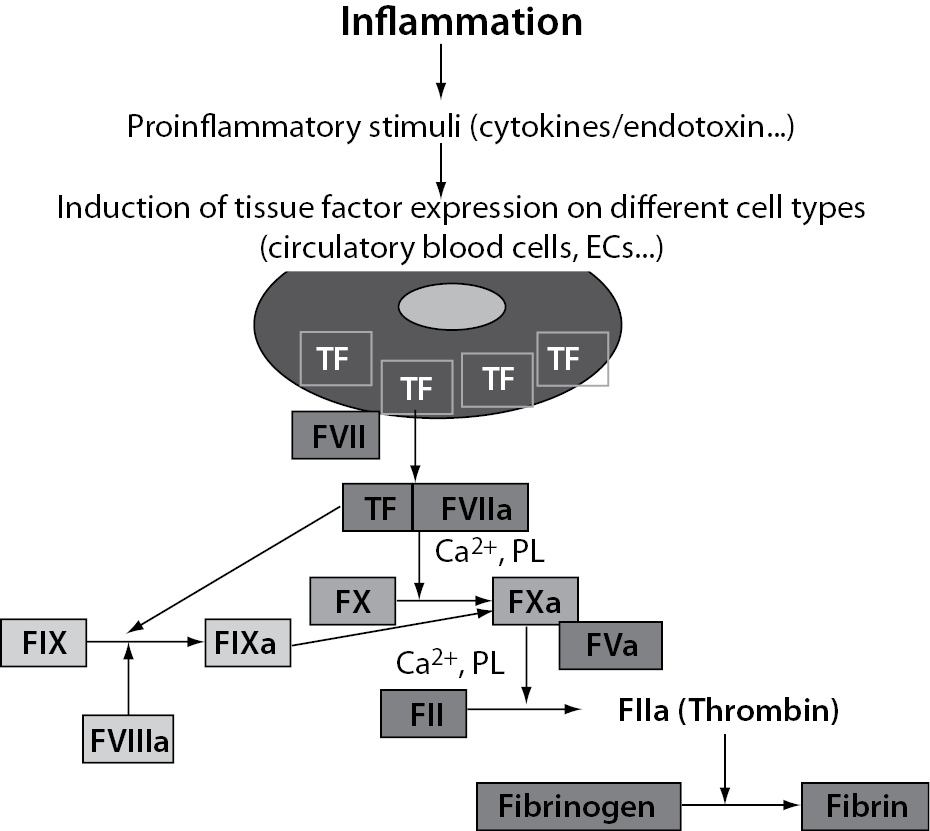
Figure 4. Tissue factor-mediated activation of coagulation induced by inflammation. TF – tissue factor; ECs – endothelial cells; PL – phospholipids.
Thrombin generated by activated coagulation cascade is a multifunctional molecule with many physiological functions. Such, thrombin has multilevel procoagulant activity. Besides it converts fibrinogen to fibrin, thrombin itself is one of the strongest platelet activators and also it is involved in feedback activation of coagulation by activating coagulation factors V, VIII, XI and XIII. Additionally, thrombin has relevant proinflammatory activity such as activation of inflammatory cells (leukocytes and ECs) with consequent increased production of inflammatory mediators and increased leukocytes adhesion and chemotaxis. Thrombin is an important regulator of cellular activation through binding to the protease-activated receptors (PARs) expressed on platelets, leukocytes and ECs. This represents the main mechanism by which activated haemostatic system augments initial inflammatory response, as it will be discussed later.
Additionally, activation of the coagulation cascade is markedly facilitated if a suitable phospholipid surface is available. Namely, coagulation reactions occur on membrane surfaces that contain negatively charged phospholipids. In normal physiological conditions, there are little negatively charged phospholipids exposed to the blood. However, both activated platelets and ECs transport their negatively charged phospholipids to the surface thus providing a phospholipid rich surface on their membrane. The phospholipid rich surface has an important role for assembly of complexes of activated coagulation factors. The presence of such a surface may accelerate several-fold the generation of thrombin and render the coagulation system less susceptible to fluid-phase protease inhibitors.
Physiologic anticoagulants in inflammation
The three major anticoagulant mechanisms involved in the regulation of coagulation activation are antithrombin (AT), PC system and tissue factor pathway inhibitor (TFPI). Antithrombin is the main inhibitor of thrombin, FXa and FIXa. PC pathway proteolytically cleaves activated coagulation cofactors FVa and FVIIIa. TFPI is the main inhibitor of the TF-FVIIa complex (Figure 5). The main function of these coagulation inhibitors is to prevent blood clotting under physiological conditions and to slow down the activated coagulation cascade after vascular injury.

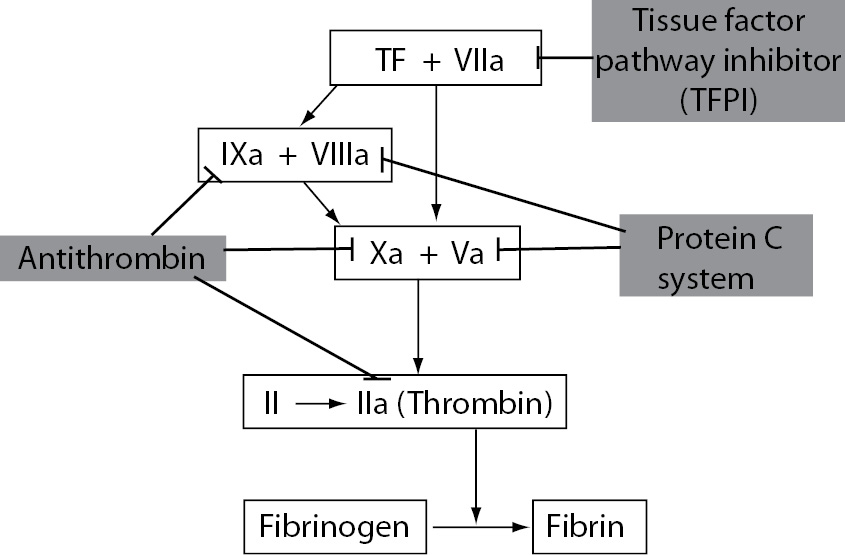
Figure 5. The three major physiological anticoagulant mechanisms: antithrombin, protein C system and tissue factor pathway inhibitor.
Normal level and function of physiologic anticoagulants appears to be important in the defence against haemostatic abnormalities in inflammatory states. There is increasing evidence that physiologic inhibitors of coagulation besides their anticoagulant actions also have important anti-inflammatory functions (26). However, the function of all three pathways can be impaired during inflammation-induced disturbance of the haemostatic system. It represents an important mechanism of procoagulant state in inflammation (27,28).
Antithrombin
In inflammatory states, the function of AT can be impaired as a result of increased consumption (due to activated coagulation cascade), decreased synthesis (as a result of a negative acute phase response) and increased degradation by proteolytic enzymes (elastase from activated neutrophils). Additionally, proinflammatory cytokines can cause reduced synthesis of glycosaminoglycans (GAGs), such as heparan sulphate on the endothelial surface, which also may contribute to the impaired AT function since endogenous GAGs act as physiologic heparin-like cofactors thus promoting anticoagulant activity of AT (29).
Some of the anti-inflammatory properties of AT are mediated by its anticoagulant actions in the coagulation cascade. By binding thrombin, AT contributes to reduced activation of proinflammatory cells (leukocytes, ECs and platelets). Further, increasing evidences suggest that AT possesses anti-inflammatory action independent of its anticoagulant activity. Such, AT directly interacts with leukocytes by binding to their receptors thus blocking the interaction of leukocytes with ECs, i.e. leukocyte migration and adhesion to ECs (30). Additionally, AT induces release of PGI2 from ECs. PGI2 acts as an inhibitor of platelet aggregation and blocks neutrophil adhering to blood vessels thus contributing to decreased production of proinflammatory cytokines and chemokines in neutrophils and ECs (31).
Protein C system
Among the three key natural anticoagulant mechanisms, the PC system appears to be the most important in regulating inflammatory response and at the same time the most negatively influenced by inflammatory states. Under physiological conditions, PC is activated by thrombin bound to endothelial cell membrane associated protein thrombomodulin (TM). Activated protein C (APC) with its cofactor protein S (PS) inactivates FVa and FVIIIa. There is increasing evidence that PC system also has important functions in modulating inflammatory response (32,33), by its anti-inflammatory and profibrinolytic activities (34,35). Anti-inflammatory actions of APC include inhibition of cytokine (TNF-a, IL-1, IL-6) production by monocytes/macrophages (36), inhibition of chemotaxis and adhesion of leukocytes to endothelium and suppression of NF-κB transcription (34). It is considered that anti-inflammatory effects of APC are mediated by the endothelial protein C receptor (EPCR) (37). Binding of APC to EPCR influences gene expression profiles of cells by blocking NF-kB nuclear translocation. This results with limited production of proinflammatory cytokines and expression of cell surface adhesion molecules in ECs (38). Further, APC has profibrinolytic action mediated by neutralization of PAI-1 activity. In addition to these anti-inflammatory actions of APC, particular components of the PC system can also contribute to attenuated inflammatory response. Besides an essential role of TM in PC system activation, it also suppresses inflammation by direct and indirect mechanisms (39). Thrombomodulin-mediated binding of thrombin attenuates proinflammatory effects of thrombin, such as platelet activation, chemotaction of monocytes and neutrophils, upregulation of leukocyte adhesion molecules and activation of inflammatory cells by binding to PARs (40). Also, by binding to thrombin, TM blocks its procoagulant action i.e., ability to clot fibrinogen. Additionally, the thrombin-TM complex activates thrombin-activatable fibrinolysis inhibitor (TAFI), an important inhibitor of fibrinolysis but also a molecule responsible for inactivation of complement component C5a, an anaphylatoxin created during complement activation (41). Considering that TM is abundantly present in the microcirculation, TAFI-mediated inactivation of C5a would be expected to protect against complement-mediated injury of the microvasculature (2).
As in case of AT, function of the PC system may be impaired because of decreased synthesis or increased consumption and degradation in inflammatory states. Furthermore, downregulation of TM and EPCR at the endothelial surface caused by proinflammatory cytokines (TNF-a, IL-1b) and neutrophil elastase from activated neutrophils may further negatively affect function of the PC system (42). In addition, increased plasma levels of C4b-binding protein (C4B-BP) as an acute phase reactant can result in a relative PS deficiency due to decreased levels of free PS, which further can contribute to a procoagulant state (6).
Impairment of the PC system appears to play an important role in the pathogenesis of sepsis and associated organ dysfunction (43,44), as it will be discussed later in the text.
Tissue factor pathway inhibitor (TFPI)
The third physiological anticoagulant mechanism is TFPI (45), a serine protease inhibitor attached to the endothelium via GAGs and secreted by ECs. As in case of AT, proinflammatory cytokines can cause reduced synthesis of GAGs on the endothelial surface, which may affect the function of TFPI. However, relatively little is known about the impact of inflammation on TFPI function. Its role in the regulation of inflammation-induced activation of haemostasis is not completely clear mostly because the majority of TFPI is associated with the vessel endothelium and direct assays of endogenous TFPI activity in vivo are not routinely available. Because of that it makes very difficult to assess whether this anticoagulant pathway is modulated by inflammation (46). Clinical experiments showed that administration of recombinant TFPI blocked inflammation-induced thrombin generation in humans (47,48). These results suggest that high concentrations of TFPI are capable of modulating TF-mediated activation of coagulation. However, the endogenous concentration of TFPI is considered not to be capable of regulating coagulation activation during inflammation.
Fibrinolytic system
Haemostasis is further controlled by the fibrinolytic system, in which the key enzyme, plasmin degrades fibrin clot. Plasmin is generated from plasminogen by activators such as tPA and urokinase plasminogen activator (uPA). The main inhibitor of these plasminogen activators is PAI-1. Binding to plasminogen activators, PAI-1 causes their inactivation thus suppressing the fibrinolytic activity.
Inhibition of the fibrinolytic system is another important component in the haemostatic disorder during inflammatory states. The initial acute fibrinolytic response in inflammatory states is a transient increase in fibrinolytic activation mediated by the immediate release of tPA from vascular ECs. However, this increased plasminogen activation is followed by delayed but sustained increase in the main fibrinolytic inhibitor PAI-1. It results with significant suppression of fibrinolytic activity and subsequent inadequate fibrin removal. The main regulators of PAI-1 activity are proinflammatory cytokines TNF-a and IL-1b (49), which induce the production of PAI-1 in vascular ECs (Figure 6). Further, platelet alpha granules also contain large amounts of PAI-1 and release it upon their activation. This may further increase the levels of PAI-1 and contribute to the fibrinolytic suppression in inflammatory states. Additionally, dysfunction of ECs induced by inflammation may also result with delayed but sustained decreased production of tPA (Figure 6).

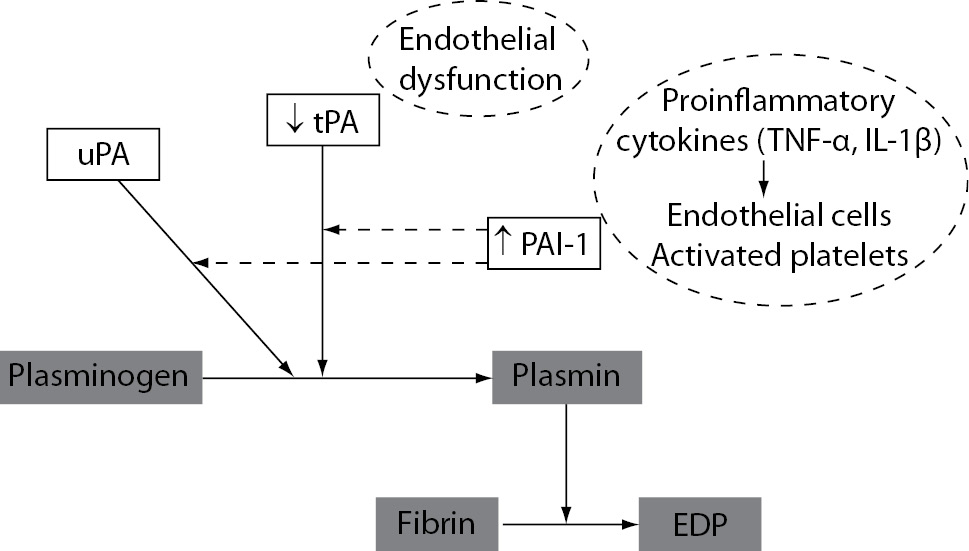
Figure 6. Mechanisms of suppressed fibrinolytic activity induced by inflammation. uPA – urokinase plasminogen activator; tPA – tissue plasminogen activator; PAI-1 – plasminogen activator inhibitor-1; FDP – fibrin degradation products.
Individual fibrinolytic components may also modulate the inflammatory response by their effect on inflammatory cell recruitment and migration. In particular, uPA and its receptor urokinase plasminogen activator receptor (uPAR) are involved in these processes. Such, uPAR expressed on leukocytes mediates leukocyte adhesion to the vascular wall thus affecting cell migration (7,50).
Mechanisms by which activated haemostatic system influences inflammatory response
The communication between inflammation and haemostasis is a bidirectional process, so activated haemostatic system also considerably modulates inflammatory activity. Individual components of the activated haemostatic system, such as activated coagulation factors thrombin, FXa and TF-FVIIa complex can directly stimulate cells involved in inflammatory response (platelets, leukocytes and ECs) with consequent increased production of proinflammatory mediators by these cells. The key mechanism by which activated coagulation factors augment inflammatory response is by their binding to PARs. The PAR family of receptors consists of four members, PAR-1 to PAR-4, which are localized on different cell types such as ECs, leukocytes, platelets, fibroblasts and smooth muscle cells. (24,51). These receptors serve as their own ligand, so that proteolytic cleavage of PAR by an activated coagulation factor activates the same receptor. Coagulation proteases activate PARs in a common way. The receptor is proteolitically cleaved at a specific site within its intracellular N-terminus. It leads to exposure a new terminus that serves as a ligand for the same receptor and results with a transmembrane signal transduction and consequent increased inflammatory response (7). PAR-1, 3 and 4 are thrombin receptors, while PAR-2 receptor can be activated by the TF-FVIIa complex or FXa. Binding of an activated coagulation factor to the individual PAR receptor results in upregulation of inflammatory responses by inducing the production of proinflammatory mediators, such as cytokines, chemokines, growth factors and cell adhesion molecules. Such, PARs seem to play an important role in linking inflammation and haemostasis (52).
Activated platelets also play an important role in enhanced inflammatory response, in particular in chronic inflammatory states such as atherosclerosis, as it will be discussed in the next section.
Further, fibrinogen as well as fibrin generated by activated coagulation cascade also mediates cellular activation in inflammation. Such, fibrinogen and fibrin are capable of recruiting and activating leukocytes by binding to the specific receptors, such as toll-like receptors (TLR) expressed at the surface of inflammatory cells (53,54). Both fibrinogen and fibrin stimulate the expression of proinflammatory cytokines (TNF-a, IL-1b) on monocytes and chemokines (IL-8, MCP-1) on ECs and fibroblasts (55).
Specific features of haemostatic disorder induced by inflammation in acute arterial thrombosis and systemic inflammatory response to infection (sepsis)
Atherosclerosis and consequent acute arterial thrombosis and sepsis represent the examples of the two clinical conditions in which close relationship between inflammation and haemostasis considerably contributes to disease severity. Because, in the next section of this article the most important features in the bidirectional relationship between inflammation and haemostasis in these two pathophysiological states will be discussed.
Relationship between inflammation and haemostasis in acute arterial thrombosis
Atherosclerosis is a chronic inflammatory process (56) in which thrombus formation on a ruptured atherosclerotic plaque is the pathological basis of an acute arterial thrombotic event such as myocardial infarction (10,57). Inflammation plays an important role in atherosclerotic process, including fatty streak formation, plaque destabilization and subsequent thrombosis (58,59). However, the mechanisms of arterial thrombosis involve not only inflammation, but rather a combination of inflammatory and prothrombotic factors that considerably affect each other (5,60). Such, platelets have an important role in the pathogenesis of arterial thrombosis including atherosclerotic plaque formation as well as its progression to acute thrombotic event (16,60). It has been shown that platelets rapidly adhere to the injured endothelium (61). In an atherosclerotic process, erosion of the vessel intimae exposes subendothelial collagen to the blood, which causes platelet adhesion, activation and subsequent aggregation (62). Once recruited to the atherosclerotic lesion, platelets become activated and express proinflammatory molecules that can additionally promote chemoattraction of leukocytes (PAF, macrophage inflammatory protein-1 (MIP-1)), stimulation of smooth muscle cells, fibroblast proliferation (PDGF, serotonin, TGF-β) and collagen synthesis. Additionally, activated platelets release proinflammatory cytokines (CD40 ligand, IL-1β) and chemokines (platelet factor-4 (PF-4), RANTES) which support monocyte recruitment to atherosclerotic plaques. Platelet adhesion to the subendothelial surface supports leukocyte rolling and extravasation through interaction of platelet P-selectin with PSGL-1 receptor expressed on leukocytes. Also, as it has already been told, activated platelets contain procoagulant molecules that directly contribute to activation of the haemostatic system.
Further, expression of the procoagulant material, in particular TF, by inflammatory cells in the unstable or ruptured plaque acts as one of the main activators of the coagulation cascade (63). Induced expression of TF on ECs, monocytes and smooth muscle cells in atherosclerotic plaques is mostly mediated by sustained increased levels of proinflammatory cytokines TNF-α and IL-1β (64). Since these cells respond to inflammatory signals within the plaque, its rupture promotes increased cell expression of TF (60,63). It has been shown that mononuclear cells in atherosclerotic plaque express much more TF than native mononuclear cells in the circulation due to sustained exposure to proinflammatory mediators in the plaque, such as cytokines (IL-1, IL-6, TNF-a) and chemokines (PDGF, MCP-1). It is apparent that circulating monocytes represent one of the principal players in the crosstalk between inflammation and activated coagulation system in an atherosclerotic process.
At the same time with an activation of the coagulation system in a progressive atherosclerotic process, there is also a reduced function of anticoagulant PC system mostly due to downregulated TM and EPCR expression in ECs overlying a plaque in coronary arteries (42). Downregulation of these two important components of the PC system is mostly mediated by sustained increased levels of proinflammatory cytokines TNF-α and IL-1β. It may result with decreased PC activation and potentially lead to more extensive thrombin generation at the site of the atherosclerotic lesion. It appears that patients with heterozygous mutation in the TM gene have a higher risk of myocardial infarction (65). As it has already been told, besides of its anticoagulant activity, TM also has anti-inflammatory actions.
Additionally, sustained increased levels of proinflammatory cytokines TNF-a and IL-1b can cause increased production of PAI-1 in ECs within the plaque. It contributes to suppressed fibrinolytic activity in atherosclerotic process.
Relationship between inflammation and haemostasis in sepsis
Another example of close interaction between immune and haemostatic system is obvious in sepsis. Sepsis is a clinical syndrome characterized by an excessive systemic host response to infection resulting with an uncontrolled activation of inflammatory response. Since the immune and haemostatic systems are tightly linked, an excessive inflammatory response can also lead to systemic activation of the haemostatic system. In fact, local activation of the coagulation in septic patients is an integral component of the host defence as an attempt to eradicate the invading microorganism. However, an exaggerated response to infection can lead to a situation in which systemic activation of the haemostatic system itself contributes to disease severity, causing a syndrome known as disseminated intravascular coagulation (DIC) (24,66). Patients with severe form of DIC may clinically present with manifest thromboembolic disease or clinically less apparent microvascular fibrin deposition that contribute to the multiple organ dysfunction (67,68). With the progression of sepsis, coagulopathy becomes increasingly more severe. Progression of the haemostatic disorder in sepsis results in the widespread formation of fibrin clots, microvascular occlusion and reduced oxygen delivery to cells and tissues with subsequent organ dysfunction (67). DIC has been shown to be an important independent predictor of organ failure and mortality in septic patients (69,70).
The majority of septic patients have coagulation abnormalities (71) ranging from subtle not clinically manifested activation of coagulation to severe coagulation disorders such as fulminant form of DIC, characterized by simultaneous microvascular thrombosis and profuse bleeding from various sites thus being a driving force in acute organ dysfunction and death (68,69,71).
The main mechanisms responsible for this severe haemostatic disorder in septic patients are mediated by excessive and uncontrolled actions of inflammatory mediators, in particular, proinflammatory cytokines. These inflammatory mediators activate coagulation system by TF-mediated pathway and at the same time significantly downregulate physiologic anticoagulant pathways (72). Thus the procoagulant activity of the haemostatic system exceeds the capacity of the physiologic anticoagulants to control the activation of coagulation. Conversely, poorly regulated activity of the haemostatic system in turn amplifies the inflammatory process causing additional organ injury.
Additionally, infectious agent itself can cause endothelial damage, which also contributes to the prothrombotic state in severe infection. The model of endotoxin-induced activation of the haemostatic system is the best studied model of the complex interactions between inflammation and haemostasis. Endotoxin is the lipopolysaccharide compound from gram negative bacteria responsible for the severe infection such as sepsis. In blood, endotoxin directly binds to CD14 receptor on monocytes and binds to ECs after complexing with lipopolysaccharide binding protein (LBP) and toll-like receptor 4 (TLR-4). Through these interactions, endotoxin induces a number of signalling pathways leading to the activation of NF-kB with consequent transcription of genes coding a number of proinflammatory mediators (73). Endotoxin also has direct positive effect on markedly increased TF expression on inflammatory cells such as leukocytes and ECs.
Further, the fibrinolytic system is directly influenced by the septic process. In most patients with sepsis, fibrinolytic activity is suppressed, while the coagulation activation still proceeds. As in other inflammatory states, the main cause of decreased fibrinolytic activity in septic patients is increased activity of PAI-1 due to sustained actions of proinflammatory cytokines.
One of the important mechanisms of disturbed regulation of the haemostatic system in septic patients is significantly reduced function of the PC system. Clinically, there is a correlation between the degree of PC reduction and mortality of septic patients (40,74). It has been shown that in patients with severe sepsis, the PC system is malfunctioning at all levels, including decreased synthesis, increased consumption and degradation and downregulation of TM and EPCR at the endothelial surface. Understanding the basic mechanisms of activation of the haemostatic system and its derangements in sepsis is crucial for the development of potential new therapeutic strategies (75). The key treatment of patients with sepsis is to treat the underlying infection with appropriate antibiotics and additional supportive treatment aimed at circulatory and respiratory support. However, abnormalities of the haemostatic system may proceed even with proper treatment. In these cases, supportive measures to manage haemostatic disorder may be considered. Since inflammation and haemostatic system are processes that function in a positive feedback loop, it is reasonable to believe that both are an appropriate target for intervention to reduce mortality in these patients. Improved understanding of the molecular mechanisms in the bidirectional interaction between inflammation and haemostasis has led to the development of new therapeutic approach for the treatment of severe sepsis. Among these agents, recombinant human activated protein C (rHuAPC) has shown the most promising results by significant reducing mortality in patients with severe sepsis (34,76). It is considered that these beneficial effects of rHuAPC can be attributed to anti-inflammatory properties in addition to its anticoagulant activity. However, although therapeutic administration of rHuAPC has been advocated in guidelines for the treatment of the severe sepsis as an additional treatment strategy (77), there is currently debate on rHuAPC efficacy since recent findings have cast some doubt on its usefulness (78-80).
Conclusion and perspectives
Improved understanding of the molecular mechanisms that play a role in the bidirectional relationship between inflammation and haemostasis could help in the clinical management of patients by identifying of new potential therapeutic targets that can modify excessive and inappropriate activation or deregulation of both systems. On the basis of experimental and clinical studies, it is likely that simultaneous modulation of both inflammatory and haemostatic activities, rather than specific therapy aimed at only one component could be more successful in the treatment of clinical states and diseases in which close link between inflammation and haemostasis considerably contribute to the pathogenesis or progression of disease. However, despite the impressive progress in understanding the molecular mechanisms linking inflammation and haemostasis during recent years, many questions remain open. Therefore, further study of the complex molecular mechanisms linking immune and haemostatic systems deserves attention of both medical experts and scientists.