Introduction
The pancreatic beta cell function and mass are decreased from the clinical onset of both types of diabetes mellitus (DM) and this is accompanied by a correspondent deterioration of glycaemic control (1). In type 1 diabetes (T1DM), the phenomenon is more severe and is mainly due to the autoimmune attack of auto reactive T cells against islet beta cells. It is assumed that about 70-90% of the beta cell mass is lost at the time of clinical presentation, which is usually abrupt, with acute metabolic decompensation (2). In type 2 diabetes (T2DM), the pathogenesis is complex, and in most cases, the reduction of beta cell function and mass is associated with different degrees of insulin resistance. The clinical presentation of patients with T2DM varies widely; from very asymptomatic to symptoms of ketoacidosis and accordingly, blood glucose concentration at diagnosis may range from mildly increased to severe hyperglycaemia (3).
In daily practice, it is sometimes difficult to discern between T1DM and T2DM because it has become apparent that they substantially overlap. Several clinical and laboratory criteria at onset, such as age, severity of symptoms, degree of hyperglycaemia, presence of ketosis, body mass index (BMI), perceived need for insulin therapy and (when possible to be measured) presence of diabetes-specific autoantibodies are used, but none of these criteria have definite thresholds that would allow a precise distinction between the two types, nor are they exclusively correlated with either type (4). In fact, it has been suggested that DM is a continuous spectrum of diseases with the autoimmune-mediated destruction of beta cells in childhood at one end and age-related metabolic deterioration at the other end of it (4).
Moreover, the “accelerator hypothesis” suggests that although on different genetic backgrounds, T1DM and T2DM are basically one and the same disorder distinguished only by the rate of beta cell destruction and the causal factors (“accelerators”) leading to beta cell loss: high intrinsic rate of apoptosis, insulin resistance and autoimmunity, which act in various degrees in different individuals (5-7). Other factors, such as incretin hormones, adiponectin or islet amyloid are also involved. It has been proposed that the accelerators are interconnected, in the sense that for example, insulin resistance may increase apoptosis (possibly through pro-inflammatory mediators and metabolic up-regulation of beta cells), followed by the release of beta cell antigens and onset of autoimmune attack in genetically susceptible subjects (5-7). Insulin resistance is central for the development of T2DM, although some patients with T1DM may also have a certain degree of insulin resistance (7,8). Insulin sensitivity decreases with age, but other factors are known to influence it: nutrients (glucose and lipids), inflammation, adipokines or disordered insulin secretion (9). Beta cell dysfunctional patterns such as delayed insulin secretory rate to a stimulus or loss of pulsatile secretion of insulin may be indicative of disease progression (5,9). Although apoptosis seem to be the main mechanism responsible for pancreatic beta cell death, studies indicate that necrosis is also implicated in the development and progression of diabetes (10).
The focus of this paper is to present the main factors and mechanisms associated with reduction of beta cell function/mass (i.e. hyperglycaemia/glucotoxicity, lipotoxicity, autoimmunity, inflammation, islet amyloid, adipokines, incretins and insulin resistance). The review also discusses methods that help assess these factors as well as beta cell function and finally, therapeutical options that may protect it by targeting the underlying factors and mechanisms with a role in disease progression.
Factors associated with progressive reduction of beta cell function/mass
Several factors are known to contribute to progressive loss of beta cell function/mass in subjects with diabetes (11). Whatever the causal factors and pathogenetic mechanisms, once the clinical onset occurs, the gradual decline of beta cell function and mass during the natural history of the disease contributes to the continuous deterioration of metabolic control.
Hyperglycemia/glucotoxicity
Pancreatic beta cells are extremely sensitive to the blood glucose concentrations and changes in its homeostasis influence beta cell function and population dynamics. Indeed, chronic exposure to abnormally high blood glucose has detrimental effects on insulin synthesis/secretion, cell survival and insulin sensitivity through multiple mechanisms (“glucotoxicity”), which in turn lead to hyperglycaemia and finally to the vicious circle of continuous deterioration of beta cell function (12). As opposed to beta cell desensitization (a temporary state of cellular refractoriness to glucose stimulation) or beta cell exhaustion (depletion of insulin stores reversible with cell rest), glucotoxicity implies irreversible changes to the cellular components of insulin production/secretion (12). It is possible however, that islet cells are in various states: desensitization, exhaustion or apoptosis in the same time. Therefore, after glucose normalization (e.g. with initiation of insulin therapy), some beta cells recover and improve their insulin secretion.
Multiple pathways and mechanisms through which chronic hyperglycaemia may impair beta cell function and cause beta cell apoptosis have been suggested (11-14):
- Prolonged exposure to increased glucose concentrations causes gradual loss of insulin gene expression, secondary to the diminished activity of the key regulators of insulin promoter activity (such as insulin gene transcriptor activators) and other beta-cell specific genes (14-17). These processes are mediated at least in part by oxidative stress.
- Beta cells exposed to an increased insulin secretory request place a high demand on endoplasmic reticulum (ER) for the synthesis of proinsulin, leading to cellular stress (18). ER stress consists of anaccumulation of unfolded proteins and triggers the unfolded protein response (UPR) in the attempt to restore the ER homeostasis (13,19). In case of severe, chronic ER stress and strong UPR, beta cell death through apoptosis (mediated by stress kinases and transcription factors) may be initiated (19). In addition to glucose, free fatty acids (FFA) and islet amyloid polypeptide (IAPP) are triggers of beta cell ER stress (19).
- Long-term sustained hyperglycemia increases the metabolic flux into the mitochondria and induces excessive generation of reactive oxygen species (ROS) which lead to chronic oxidative stress (14). Clinical studies indicated that diabetic patients are exposed to chronic oxidative stress and present increased pro-oxidants and markers of oxidative tissue damage (20,21). Moreover, studies on islets isolated from pancreata of subjects with T2DM showed increased markers of oxidative stress and these correlated with the degree of glucose-stimulated insulin secretion (GSIS) impairment (22). In hyperglycaemic states, excessive ROS production is generated by mitochondrial oxidative phosphorylation during anaerobic glycolysis and from alternative pathways into which glucose is shunted when the glycolytic capacity is exceeded: glucose autooxidation, non-enzymatic glycation, protein kinase (PK)C activation, and from the α-ketoaldehyde, hexosamine and sorbitol pathways (14,15). The oxidative stress activates stress-induced pathways that damage the beta cells by inducing defective insulin biosynthesis and secretion, and ultimately apoptosis (11,23). Elevated ROS disturb the integrity and function of cellular proteins (e.g. enzymes, receptors, transport proteins), lipids and deoxyribonucleic acid (DNA): they degrade polyunsaturated fatty acids of the membranes, induce lipid peroxidation and aminoacids oxidation and damage purine and pirimidine bases (24). The apoptotic and cell death mechanisms are also induced by disruption of mitochondrial membrane integrity and release of cytochrome c from the outer membrane and by accumulation of mitochondrial DNA mutations (24,25). Recent data indicate that the oxidative stress and ER stress are closely linked in the sense that cellular ROS can increase the misfolded protein accumulation in the ER, which amplifies ROS production that in turn further increases the ER stress, disrupt insulin production, and initiate beta cell death (25,26). It should be noted that the strong, continuous activation of stress signaling pathways results in beta cell dysfunction and apoptosis, while the temporary, fluctuating and probably less severe activation may promote adaptation and cell survival (24,25). Finally, the oxidative stress might also be involved in the hyperglycaemia-induced insulin resistance, which impacts on beta cell function (27).
- Data indicates that chronic hyperglycaemia leads to decreased number of mitochondria and changes of their morphology (increased volume and outer surface area, reduction of proteins in the inner membrane, increased variability in mitochondrial size) in the beta cells. These changes in morphology and function are associated with impaired GSIS through impaired oxidative phosphorylation, decreased mitochondrial Ca2+ capacity and decline in ATP generation (28,29). In addition, disruption in Ca2+ homeostasis (e.g. changes in glucose-induced Ca2+ influx,ER Ca2+ depletion) negatively impacts on beta cell growth/function and on the insulin secretion pathway (30).
- Evidence exists that chronic hyperglycemia induces non-immune mediated inflammatory pathways (e.g. production of interleukin (IL)-1β, nuclear factor-kB (NF-kB) and Fas receptor) in islets, although this was not confirmed in other studies (13,18,31). It is argued that pro-inflammatory signals triggering apoptosis are related to mechanisms associated with oxidative and ER stress (13,18,31).
Lipotoxicity
Diabetes is often associated with changes in lipoprotein profiles and increased FFA concentrations (11). Prolonged exposure to increased FFA levels (derived from adipocyte lipolysis or from lipoprotein hydrolysis) may have negative effects on beta cell function (i.e. attenuate GSIS) and lead to accumulation of toxic FA metabolites in the islet cells (“lipotoxicity”) (13,15,32). Several observations suggest that the FFA toxicity on beta cells depends on several parameters (concentration of unbound FFA, level of unsaturation, nature of the isomers or chain length) (33).
However, there are reports (mainly coming from in vitro experiments and some animal studies) indicating that the deleterious effects of lipids on beta cells occur only in the presence of hyperglycaemia, when elevated FA are not oxidized in mitochondria and are shunted towards the esterification pathways, with accumulation of long-chain acyl-CoA esters in the cytoplasm (“glucolipotoxicity”) (34,35).
Several mechanisms by which FA may affect beta cell function and survival have been suggested (15). The accumulation of intermediates of lipid metabolism may affect beta cells directly or indirectly, through lipid-derived signals (13). Prolonged exposure to high FA concentrations in hyperglycaemic conditions results in decreased insulin gene expression (15). The impairment of insulin secretion may also result from mechanisms related to modulation of G protein-coupled receptor 40 and increased expression of uncoupling protein-2 (15).
Although most of the research has been focused on the effects of FFA on beta cell function, emerging data suggest that cholesterol metabolism/lipoprotein fractions may also play a role in the progression of beta cell failure. Low density lipoprotein (LDL) and very low density lipoprotein particles were shown to induce apoptosis, oxidized LDL reduce preproinsulin expression levels in isolated beta cells, while high density lipoprotein (HDL) particles are protective (36,37). These effects were at least in part mediated by c-Jun N-terminal kinase (JNK) and caspase-3 pathways (36,37). Recent work has indicated that the disruption of cholesterol transport by reduced function of cholesterol transporters results in sterol accumulation, islet inflammation, and impairment in GSIS (38). Thus lipoproteins seem to be important modulators of beta cell function/survival (39).
Autoimmunity and inflammation
The role of autoimmunity in T1DM has been long recognized. During the progression of the disease, autoimmune-mediated apoptosis of beta cells leads to a continuous reduction of insulin secretion and beta cell mass (5). Some phenotypic T2DM subjects also present markers of autoimmunity and these are correlated with beta cell function, suggesting a possible role of immune system in the pathogenesis of T2DM (4,40,41). Reports have indicated that about 10% of individuals with T2DM have diabetes-specific autoantibodies, but the percentage is higher in younger-age and in leaner groups (42). More recent data indentified T2DM subjects with islet-reactive T cell responses (with or without autoantibodies) and these were correlated with a lower beta cell function (41).
Although the mechanisms behind autoimmunity and the trigger factors might be different, immune-cell infiltration of pancreatic islets and increased local cytokine production (insulitis) have been described in both types of DM (43). The inflammatory processes in the islets share common final signaling pathways and effector molecules (43). Apoptosis is induced by cytokines, stimulation of Fas expression on beta cells which renders them more sensitive to T-cell mediated killing via Fas-Fas ligand (mainly in T1DM) or by activation of NF-kB and mitogen-activated protein kinase (MAPK)/JNK pathways (both types) (43).
Islet cells are metabolically and immunologically up-regulated when functionally stressed by hyperglycemia (44). A pathogenetic mechanism that leads to islet inflammation in T2DM begins with chronic metabolic stress (high levels of blood glucose and FFAs), which induces an inflammatory response in pancreatic islets, consisting of increased production of cytokines and chemokines (45). Among them, a crucial role seems to be played by IL-1β, a master cytokine that regulates many other pro-inflammatory cytokines (such as tumor necrosis factor α(TNFα), IL-6) and chemokines (like chemokine (C-X-C motif) ligand (CXCL) 1, monocyte chemotactic protein (MCP)-1, macrophage inflammatory protein (MIP) α) (45). By enhanced production of cytokines, cytotoxic factors and chemokines and local attraction of macrophages, IL-1β contributes to apoptosis and beta cell function impairment (45). Other studies have identified CXCL10 to play crucial role in triggering beta cell apoptosis, and elevated levels of serum CXCL10 have been found in both T1DM and T2DM (46).
On the other hand, adipose tissue-derived factors (pro-inflammatory adipokines), discussed later, have local and systemic effects on metabolism and contribute to the chronic inflammatory process (47). In addition, adipocytes may present antigens that activate macrophages and other immune cells in the adipose tissue, and thus play a role in inflammation and autoreactive responses (40).
Not only that low-grade systemic inflammation has been shown to induce beta cell dysfunction, but chronic inflammation also triggers beta cell death and is a risk factor for autoimmunity (40,48). Apoptotic beta cells may themselves present antigens and stimulate the development of an autoimmune response and associated inflammatory islet lesions (40).
Thus, islet autoimmune processes occur in both types of DM, although differences exist: the cell-mediated autoimmune disease in T1DM is rather triggered by environmental factors, while in T2DM it seem to result from chronic inflammatory responses associated with obesity and metabolic stressors (40).
Adipokines
Adipokines are hormone-like peptides/proteins released from adipose tissue with a role in the “adipo-insular” axis: some (such as leptin) act as pro-inflammatory cytokines and contribute to beta cell failure, while others have protective effects on beta cell function and survival (49).
It has been shown that leptin may induce inflammatory reactions and apoptosis upon chronic exposure. Leptin exerts direct effects on pancreatic beta cells that express leptin receptors, where it activates multiple signaling pathways leading to inhibition of GSISand decreased pre-proinsulin levels (49). Other cytokines released by adipocytes (e.g. IL-6 and TNFα) may also modulate beta cell survival, although it has not been clarified if the quantities released in circulation are enough to cause beta cell dysfunction. One recent report indicated the TNFαincrease the expression of amylin (and not proinsulin) in beta cells (50). Newer adipocyte-derived secretory molecules, like resistin and apelin, have been shown to reduce insulin secretion.
Adiponectin, on the other hand, is structurally related with TNFα, but has a number of beneficial effects: it exerts insulin sensitising actions (mainly in the liver), promotes beta cell function and survival, has local and systemic anti-inflammatory effects (51). Under detrimental metabolic conditions, adiponectin is down-regulated, resulting in low plasma concentrations (51). At the level of beta cells adiponectin acts through its receptors and appears to increase the GSIS (52). The major effect on pancreatic beta cells seems however to be the anti-apoptotic one, by activation of pro-survival kinases, reduction of intracellular ceramide levels and increase of sphingosine-1 phosphate (an anti-apoptotic metabolite) concentrations (53).
Another adipokine, visfatin, originally presented as an insulin mimetic, has been shown to enhance insulin secretion and beta cell proliferation and to inhibit beta cell apoptosis, possibly through regulation of pancreas duodenum homeobox-1 (PDX-1) expression (49).
Islet amyloid
Hypersecretion of IAPP and amyloid deposits (consisting mainly of IAPP aggregates) in the pancreatic islets have been considered a pathological characteristic of T2DM and its role in the beta cell death has long been researched (54,55). IAPP is a normal product of beta cells co-secreted and stored with insulin in secretory granules (55). Its functions are not fully understood, but it seems that it limits the glucose-stimulated insulin secretion (although a stimulatory effect on basal insulin secretion has also been described) and has an inhibitory effect on gastric emptying and appetite (54,55).
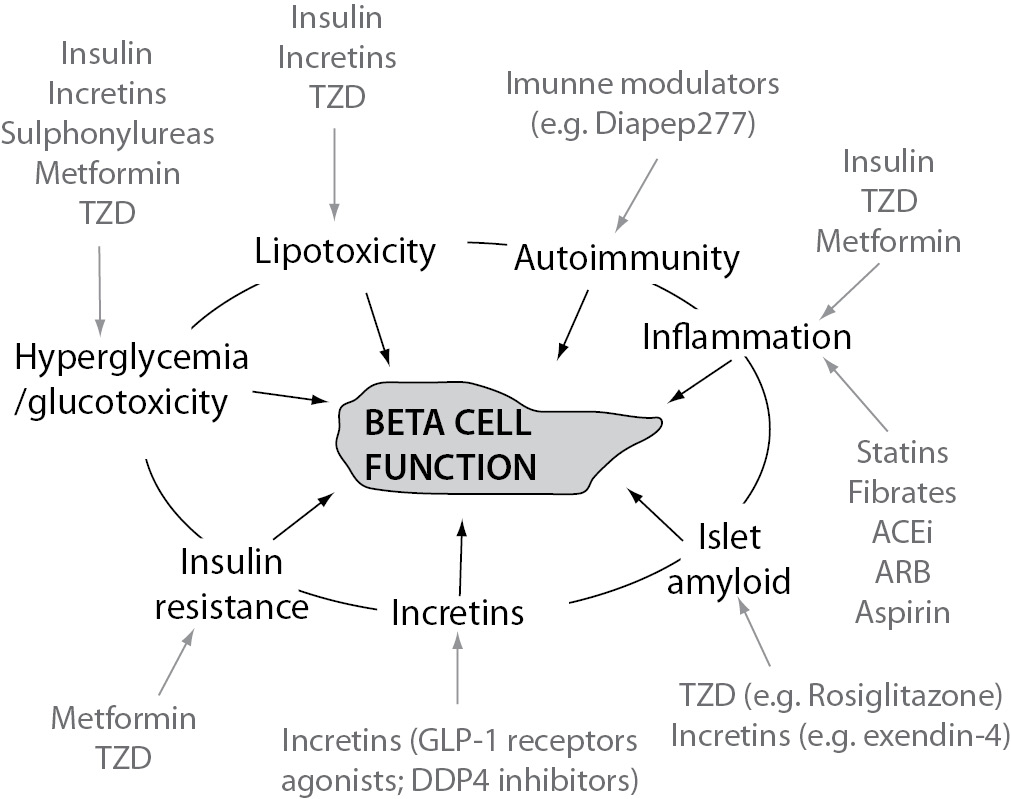
Figure 1. Main factors associated with progressive alteration of beta cell function and pharmacologic agents that influence them. TZD - thiazolidindione; GLP-1 - glucagon-like peptide-1; DPP4 - dipeptidyl peptidase-4; ACEi - angiotensin converting enzyme inhibitors; ARB - angiotensin receptor blocker
In order for IAPP to aggregate it has to assume a β-sheet conformation (56). Human IAPP first precipitates and forms amorphous aggregates that finally assemble into fibrils (56). Although the aggregation starts intracellularly, most of amyloid fibrils are found in the extracellular space where they form deposits (54,56). A high secretory demand may result in an inappropriate protein folding (in the ER), processing (in the secretory granules) and/or trafficking into the beta cells (54). Insulin (but not proinsulin) molecules seem to form heteromolecular complexes that stabilize IAPP and inhibit fibril formation, and thus an increased proinsulin/insulin ratio seen in diabetes might affect fibril formation (56). It has also been implied that in order for fibrils to form, a series of changes of the cellular environment should occur: in pH, temperature, salt/calcium concentrations, lipidoxidation, glycation, oxidation, deamidation of peptides (56). It has also been suggested that increased glucose and lipid concentrations enhance amyloid fibril formation (54).
The mechanisms by which amyloid formation is related to beta cell dysfunction and death are not fully understood. Studies indicated that the small oligomers and larger fibrils are more cytotoxic (56,57). These fibrils may insert into the lipid layer of cell membrane and form pores (ion channel-like structures) that change the ion flux or cause a non-specific membrane disruption (55,57). Destabilization of intracellular ion environment and generation of ROS may disrupt the normal insulin secretory kinetics and trigger beta cell death (54). Recent findings suggest that beta cell apoptosis and death is induces by aggregated IAPP that activate the production of IL-1β and through induction of ER stress and UPR (55,58). Nevertheless, a strong correlation between islet amyloid deposition and reduced beta cell area and increased beta cell apoptosis has been described in T2DM (59).
Incretins
The positive effects of incretin hormones on beta cell function and mass are known (60,61). Through binding on specific receptors on beta cells and increases of intracellular cyclic AMP (cAMP) and calcium, incretins stimulate the GSIS, improving both first- and second-phase responses and restoring the biphasic profile (60,62,63). They also up-regulate several beta cell-specific genes, including proinsulin, glucokinase and glucose transporters (60,64). Because the stimulation of insulin synthesis occurs by up-regulation of insulin transcription and translation and stabilization of insulin mRNA, the beta cell secretory capacity and insulin stores are preserved (63-65). It has also been shown that glucagon-like peptide-1 (GLP-1) prevents glucolipotoxicity in beta cells via PKB activation (66). Through these effects incretins protect and improve the function of beta cells. Moreover, the chronic effects of incretin hormones seem to favor the maintenance of beta cell mass, by stimulating the regeneration, proliferation and neogenesis while enhancing resistance to apoptosis, as indicated by animal and in vitro studies (67-69). The mechanisms behind the positive effects on pancreatic beta cell preservation are not fully elucidated, but data indicate that cAMP/PKA, phosphatidylinositide 3-kinases (PI-3K)/PKB pathways and PDX-1 transcription factor are involved (69).
In T2DM there is both an impairment of incretin release and a resistance to their action, which contributes significantly to insulin deficiency and beta cell dysfunction (70). Hyperglycemia further amplifies the alteration of the incretin effect, in part by down-regulating or de-sensitization of their specific receptors (71).
Although the exact mechanisms behind the impaired incretin responsiveness in diabetes are not fully understood, it has been suggested that receptor mutations, down-regulation or de-sensitization, post-receptor defects of intracellular machinery or reduction of beta cell function/mass per se might play a role (72-74). In addition, several factors, such as high BMI and high glucagon levels seem to be associated with a decreased GLP-1 secretory response (75).
Since the incretin activity is defective in subjects with diabetes, it can be hypothesized that their pro-survival effects on beta cells are also reduced and this may contribute to the beta cell demise.
Insulin resistance
There is a large body of literature emphasizing that insulin resistance places an augmented demand on beta cells to increase the insulin secretion in order to compensate the defect in insulin action, and this leads to a progressive beta cell dysfunction and failure (76). This will not be discussed in detail here. The mechanisms by which insulin resistance contributes to beta cell failure are not fully elucidated. One hypothesis is that insulin hypersecretion finally causes beta cells exhaustion, while other suggests that the factors that lead to insulin resistance (e.g. lipotoxicity) also result in beta cell failure (76).
The role of insulin resistance in T2DM is well known, but it has become apparent that it also occurs in subjects with T1DM, at the level of skeletal muscle and liver (8). Recent studies indicate that the degree of insulin resistance in adults with T1DM is independent of glycemic control (77). The mechanisms of insulin resistance in T1DM at the level of skeletal muscle are similar with those in T2DM, but in the liver they seem to be somewhat different (8).
Assessment of beta cell function and main pathogenetic mechanisms
Since functional beta cell mass cannot be directly measured in patients with diabetes, methods that evaluate the beta cell function are used instead. This evaluation in physiologic conditions is however difficult because it is interrelated with other variables, mainly insulin sensitivity (78). Changes in insulin sensitivity (e.g. insulin resistance augmentation with increased body weight) demand compensation through a proportionate adjustment of insulin secretion by beta cells in order to maintain glucose homeostasis (79,80). In fact, it was proven that between insulin secretion and insulin sensitivity there is a non-linear, hyperbolic relationship, and the product of the two variables is constant (“disposition index”) (80,81). Therefore, a correct evaluation of beta cell function requires quantification of insulin secretion in relation with insulin sensitivity (evaluation of the disposition index) (78,82).
In addition, the problem of hepatic extraction and non-constant clearance of insulin has to be considered. For this reason, C peptide values could be measured instead, because C peptide is co-secreted in equimolar amounts with insulin, undergoes non-significant hepatic extraction, has a relatively constant kinetics and thus reflects more accurately the pancreatic insulin secretion (83). Moreover, a good assessment of beta cell function requires an evaluation of the dynamic insulin secretory response to a stimulus, which, if not standardized, needs an appropriate method of normalization (83). It should be mentioned though that the normalization methods necessitate a specialized software and expertise (83).
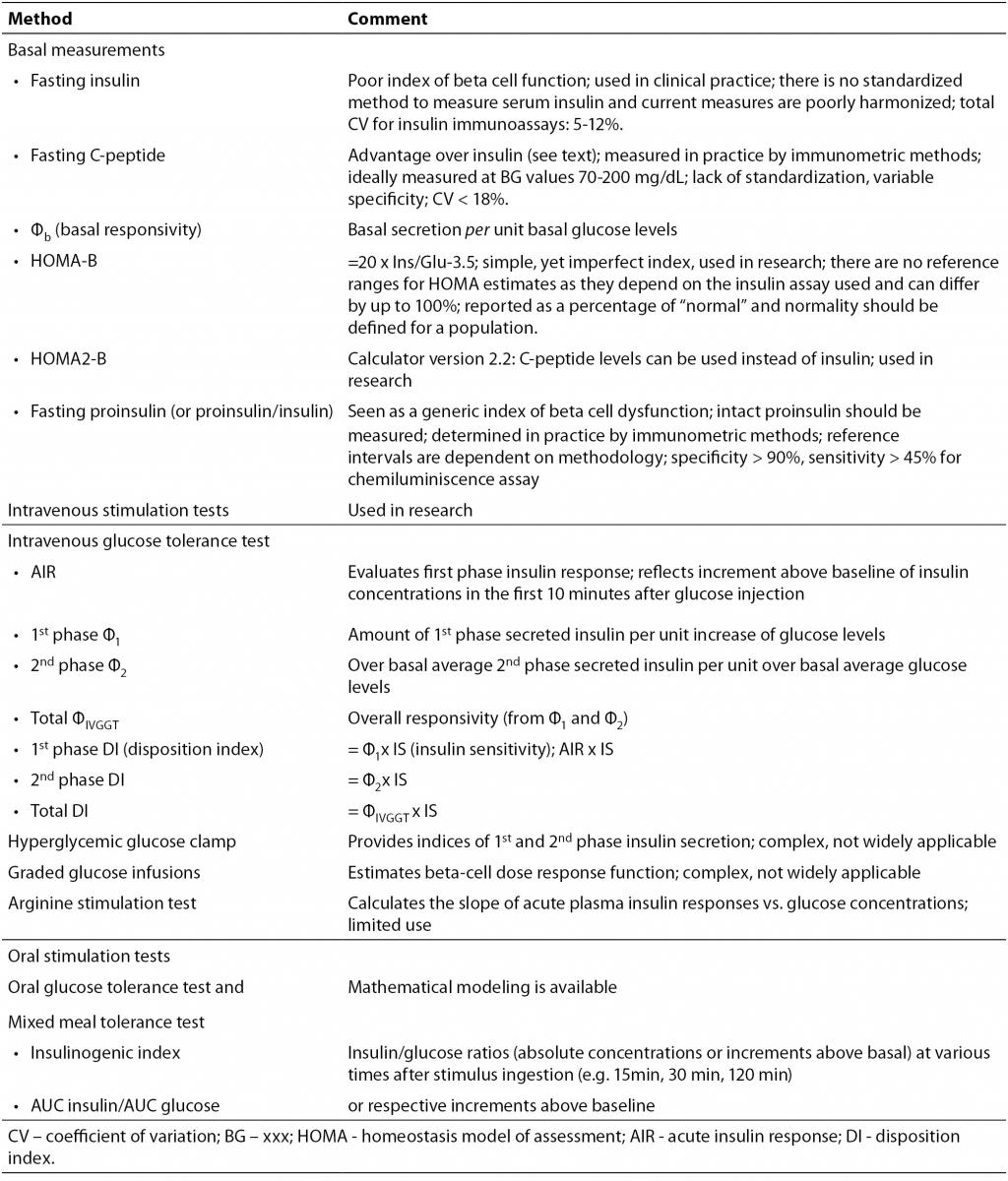
The gold-standard methods for evaluation of insulin secretion and sensitivity are considered hyperglycemic and euglycemic-hyperinsulinemic clamp, respectively, but they have many inconveniences (high cost, time consuming, complicated technique requiring trained personnel) which make their use in clinical practice difficult (even for some research purposes) (82,84). Therefore, simpler methods are used in practice, such as oral stimulation tests (with glucose or mixed meals), intravenous stimulation tests (with glucose, glucagon or arginin) or simply surrogate markers derived from basal measurements (such as homeostasis model assessment (HOMA) indexes) (Table 1) (78,85,86). Complex mathematical analyses have been developed to model the parameters obtained and extract multiple indexes that allow a better interpretation of the tests (85,86).
Table 1. Surrogate methods for evaluation of beta cell function (78,86,88).
The tests that evaluate beta cell function and insulin sensitivity may be used for a better understanding of the fundamental mechanisms that interconnect the two variables and to identify the factors that signal beta cell adaptation to changes of insulin sensitivity (82). Some possible candidates have been suggested (such as glucose, lipids, incretins, pancreatic polypeptide as a marker of vagal activity), but their relative contributions have not been yet exactly quantified (82). In addition, the signals from beta cells that might regulate insulin sensitivity should be examined in depth (82).
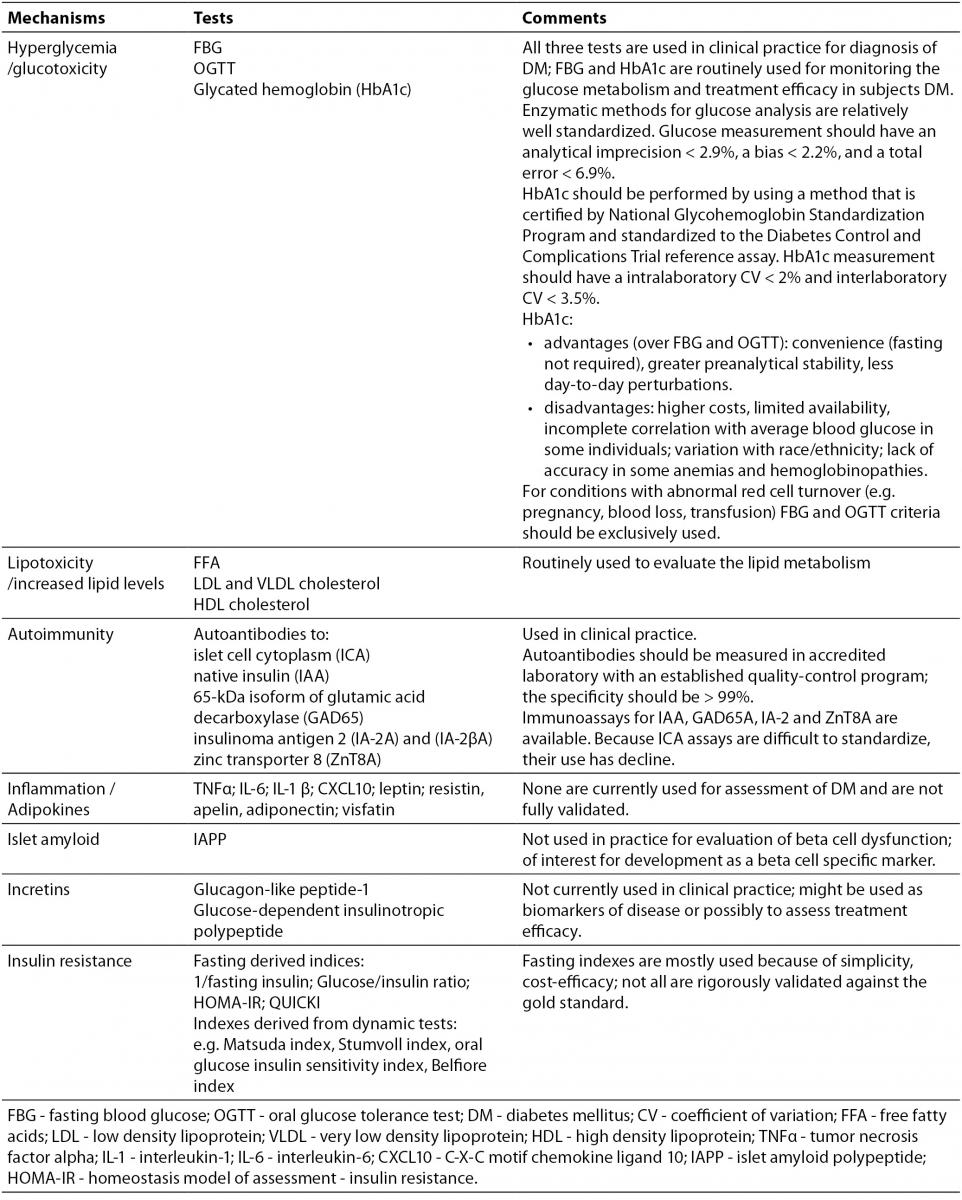
In order to better characterize and understand the main pathogenetic mechanisms that contribute to disease development in a certain individual with DM, additional markers could be used, apart from those that evaluate beta cell function. A panel of such serological biomarkers is suggested in Table 2. These tests might be useful in indentifying, characterizing and segregating different patient subtypes presenting some (or all) of the pathogenetic factors and who might benefit from appropriate therapeutical interventions that target specific mechanisms (discussed below). Some of these markers are already used in clinical practice (e.g. markers of glucose metabolism, lipids, diabetes-specific autoantibodies), others are used in research or are under development.
Table 2. Serological markers that evaluate main pathogenetic mechanisms involved in DM onset and progression (see text for details) (87,88).
Clinical considerations
Individuals with diabetes and similar clinical features may have different molecular basis that influence the rate of disease progression. Therefore, a better characterization of the underlying defects in individuals with this disease is needed and it is important that they are taken into consideration when the treatment strategy is chosen. One approach for beta cells protection is to target the underlying mechanisms/factors that are deleterious for their function.
A number of pharmacological agents and treatment algorithms are now available for the management of hyperglycemia (76,89). Glucotoxicity can be ameliorated if hyperglycemia is reduced. This is obtained by improvement of peripheral glucose uptake (with metformin or thiazolidindione (TZD)), reduction of hepatic glucose production (with metformin, TZD, incretins), improvement of insulin secretion (with sulphonylureas (SU) or incretins), exogenous administration of insulin (76). Moreover, some of the agents (TZD, insulin, incretins) have direct protective effects on beta cell function (11,76). Both TZDs and incretins have been shown to decrease beta cell apoptosis, promote beta cell proliferation, increase insulin synthesis and secretion and improve the insulin secretion pattern (61,76). Some studies indicated beneficial effect of early insulin therapy on beta cells in patients with latent autoimmune diabetes of adults (LADA) (90). Similarly, early intensive insulin therapy with multiple injections or continuous subcutaneous insulin infusion in subjects with newly diagnosed T2DM has favorable outcomes in terms of recovery and maintenance of beta cell function (especially restoration of acute insulin response) (91). The exact mechanisms behind the beneficial effects of exogenous insulin administration on beta cells are not completely elucidated, but it is though that it allows cells rest, at least in part by down-regulating their metabolism and/or by releasing them from the hyperglycemic stress (92). Insulin therapy may also protect pancreatic beta cells by decreasing the severity of insulitis, suppressing the inflammatory processes, reducing antigen expression and subsequent amelioration of T cell responses/number of infiltrative cells in the islets (93). Prolonged exposure to SU on the other hand has been associated with beta cell exhaustion, desensitisation and possibly acceleration of oxidative stress and apoptosis, leading to a progressive reduction of insulin production capacity and deterioration of glycemic control over time (94).
TZDs and insulin are also potent suppressors of lipolysis and thus reduce the amount of FFA released in circulation (76). TZD also mobilize the fat out of beta cells, liver and muscle (76).These processes ameliorate lipotoxicity. Incretins have also been shown to correct glucolipotoxicity, by reducing glucose and FFA levels or possibly though mechanisms related to PKB activation and NF-kB pathway (95).
The anti-inflammatory effects of insulin have already been mentioned, but these have been also demonstrated for TZDs and metformin, as well as for other (non-hypoglycemic) drugs that are used by subjects with diabetes (e.g. statins, fibrates, angiotensin converting enzyme inhibitors, angiotensin receptor blockers and even high-dose aspirin) (96,97).
With regards to immune interventions targeted towards autoimmunity and aimed to protect remaining insulin-producing beta cells and their function, these have been used only in clinical trials of T1DM (and some of LADA), but unfortunately, the promising results of phase 2 studies could not be confirmed in phase 3 trials for all agents (98).
So far, there is limited literature regarding agents that would ameliorate islet amyloid deposition and its toxic effects on beta cells, although some recent reports suggest that rosiglitazone (a TZD) and exendin-4 (a GLP-1 receptor agonist) may have protective effects via PI-3K/AKT pathway (99). The role of incretin therapy in T2DM and the positive effects of incretins on beta cells have been extensively reviewed elsewhere (61,72,76). Finally, insulin sensitivity is improved by agents that have potent insulin sensitizing effects (metformin, TZD), but possibly the reduction of hyperglycaemia per se may also improve it (76).
In addition to drug therapy, other therapeutical options like intensive lifestyle intervention and gastric bariatric surgery (by Roux-en-Y gastric bypass method)result in decrease of body weight and improvement of beta cell function, while pancreatic islet transplantation increase the insulin-secreting cell mass (100).
All these pathophysiological aspects should be considered when choosing the treatment intervention, in order to better protect and preserve beta cells and obtain long-term benefits. Possibly, a combination of two/more agents that concomitantly target more of these mechanisms/factors would render most benefits.
Conclusions
This paper highlights the heterogeneity of diabetes and the pivotal role of beta cell dysfunction. It indicates the main factors that influence beta cell function altering the natural course of the disease. A correct assessment of beta cell function requires concomitant evaluation of insulin sensitivity, because the two variables need to be judged in relation to each other. In any case, a better understanding of the fundamental mechanisms that connect the two processes is necessary. Clinical considerations are made regarding the therapeutical options which impact on various factors/mechanisms that contribute to the progressive loss of beta cell function/mass.