References
1. Andrews NC. Medical Progress: Disorders of Iron Metabolism. N Engl J Med 1999;341:1986-95.
3. Liu K, Kaffes AJ. Iron deficiency anaemia: a review of diagnosis, investigation and managment. Eur J Gastroenterol Hepatol 2012;24:109-16.
4. Yen AW, Fancher TL, Bowlus CL. Revisiting hereditary hemochromatosis: Current Concepts. Am J Med 2006;119:391-9.
5. Stankowski JN, Dawson VL, Dawson TM. Ironing out tau's role in Parkinsonism. Nat Med 2012;18:291-5.
6. Lei P, Ayton S, Finkelstein DI, Spoerri L, Ciccotosto GD, Wright DK. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat Med 2012;18:291-5.
7. Huang X, O`Brien PJ, Templeton DM. Mitochondrial involvement in genetically determined transition metal toxicity I. Iron toxicity. Chem Biol Interact 2006;163:68-76.
8. Guidi GC, Santonastaso CL. Advancements in anemias related to chronic conditions. Clin Chem Lab Med 2010;48:1217-26.
9. Andrews, NC. Metal transporters and disease. Curr Opin Chem Biol 2002;6:181-6.
10. Latunde-Dada GO, Simpson JR, Mckie AT. Recent advances in mammalian haem transport. Trends Biochem Sci 2006;31:182-8.
11. Papanikolaou G, Pantopoulos K. Iron metabolism and toxicity. Toxicol Appl Pharmacol 2005;202:199-211.
12. Aisen P, Enns C, Wessling-Resnick M. Chemistry and biology of eucariotic iron metabolism. Int J Biochem Cell Biol 2001;33:940-59.
13. Watt RK. Oxido-reduction is not the only mechanism allowing ions traverse the ferritin protein shell. Biochim Biophys Acta 2010;1800:745-59.
14. Puntarulo S. Iron, oxidative stress and human health. Mol Aspect Med 2005;26:299-312.
15. Kurz T, Gustafsson B, Brunk UT. Cell sensitivity to oxidative stress is influenced by ferritin autophagy. Free Radic Biol Med 2011;50:1647–58.
16. Chasteen ND, Harrison PM. Mineralization in ferritin: An efficient means of iron storage. J Struct Biol 1999;126:182-94.
17. De Domenico I, Ward DM, Kaplan J. Specific iron chelators determine route of ferritin degradation. Blood 2009;114:4546-51.
18. Asano T, Komatsu M, Iwai YY, Ishikawa F, Mizushima N, Iwai K. Distinct mechanisms of ferritin delivery to lysosomes in iron-depleted and iron-replete cells. Mol Cell Biol 2011;31:2040-52.
19. Surguladze N, Thompson KM, Beard JL, Connor JR, Fried MG. Interaction and reactions of ferritin with DNA. J Biol Chem 2004;279:14694-702.
20. Alkhateeb A, Connor JR. Nuclear ferritin: A new role for ferritin in cell biology. Biochim Biophys Acta 2010;1800:793-7.
21. Bou-Abdallah F, Santambrogio P, Levi S, Arosio P, Chasteen ND. Unique iron binding and oxidation properties of human ferritin: A comparative analysis with human H-chain ferritin. J Mol Biol 2005;347:543-54.
22. Nie GN, Chen G, Sheftel AD, Pantopoulos K, Ponka P. In vivo tumor growth is inhibited by cytosolic iron deprivation caused by the expression of mitochondrial ferritin. Blood 2006;108:2428-34.
23. Richardson DR, Lane HJR, Becker E, Huang MLH, Whitnhall M, Rahmanto YS, et al. Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc Natl Acad Sci U S A 2010;107:10775-82.
24. Santambrogio P. Over-expression of mitochondrial ferritin affects the JAK2/STAT5 pathway in K562 cells and cause mitochondrial iron accumulation. Haematologica 2011;96:1424–32.
25. Choen LA. Serum ferritin is derivied primarly from macrophages through a nonclassical secretory pathway. Blood 2010;116:1574-84.
27. De Domenico I, Vaughn MB, Paradkar PN, Lo E, Ward DM, Kaplan J. Decoupling ferritin synthesis from free cytosolic iron results in ferritin secretion. Cell Metab 2011;13:57–67.
28. Wang RH, Li C, Xu X, Zheng Y, Xiao C, Zerfas P, et al. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab 2005;2:399-409.
29. Fisher J, Devraj K, Ingram J, Slagle-Webb B, Madhankumar AB, X Liu, et al. Ferritin: A novel mechanism for delivery of iron to the brain and other organs. Am J Physiol Cell Physiol 2007;293C641-9.
30. Han J, Seaman WE, Di X, Wang W, Willingham M, Torti FM, et al. Iron uptake mediated by binding of H-ferritin to the TIM-2 receptor in mouse cells. PLoS ONE 6: e23800.
31. Cavill I. Iron and erythropoietin in renal disease. Nephrol Dial Transplant 2002;17:19-23.
32. Finch C. Regulators of iron balance in humans. Blood 1994;6:1697-702.
33. Ozaki M, Awai T, Kawabata M. Iron release from haemosiderin and production of iron-catalysed hidroxyl radicals in vitro. Biochem J 1988;250:589-95.
34. Umbreit J. Iron deficiency: A Concise Review. Am J Hematol 2005;78:225-31.
35. Munoz M, Garcia-Erce JA, Remacha AF. Disorders of iron metabolism. Part 1: molecular basis of iron homeostasis. J Clin Pathol 2011;64:281-6.
36. Conrad ME, Umbreit JN. Pathways of iron absorption. Blood Cells Mol Dis 2002;29:336-55.
37. McKie AT, Barrow D, Latunde-Dada GO, Rolfs A, Sanger G, Mudaly E, et al. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science 2001;291:1755-9.
38. Atanasova B, AC Li, Bjarnason I, Tzatchev KN, Simpson RJ. Duodenal ascorbate and ferric reductase in human iron deficiency. Am J Clin Nutr 2005;81:130-3.
39. Ohgami RS, Campagna DR, McDonald A, D Fleming. The Steap proteins are matalloreductases. Blood 2006;108:1338-94.
40. Canonne-Hergaux F, Gruenhied S, Ponka P, Gross P. Cellular and subcellular localisation of the Nramp2 iron transporter in the intestinal brush border and regulation by dietary iron. Blood 1999;93:4406-17.
41. Garric MD, Singleton ST, Vargas F, Kuo HC, Zhao L, Knopfrl M. DMT1: Which metals does it transport? Biol Res 2006;39:79-85.
42. Conrad ME, Umbreit EG, Moore LN, Porubcin M, Hainsworth MJ, Simovich MT, et al. Separate pathways for cellular uptake of ferric and ferrous iron. Am J Physiol Gastrointest Liver Physiol 2000;279:767-74.
43. Umbreit JN, Conrad, ME, Hainsworth LN, Simovich M. The ferrireductase paraferritin contains divalent metal transporter as well as mobilferrin. Am J Physiol Gastrointest Liver Physiol 2002;282:534-39.
44. Ranganathan PN, Lu Y, Fuqua BK, Collins JF. Immunoreactive hephaestin and ferroxidase activity are present in the cytosolic fraction of rat enterocytes. Biometals 2012 [Epub ahead of print].
45. Simovich MJ, Conrad ME, Umbreit JN, Moore EG, Hainsworth LN, Smith HK. Cellular localisation of proteins related to iron absorption and transport. Am J Hematol 2002;69:164-70.
46. Hou S, Reynolds MF, Horrigan FT, Heinemann SH, Hoshi T. Reversible binding of heme to proteins in cellular signal transduction. Acc Chem Ress 2006;39:918-24.
47. Latunde-Dada GO, Takeuchi K, Simpson RJ, McKie AT. Haem carrier protein 1 (HCP1): Expression and functional studies in cultured cells. FEBS Lett 2006;580:6865-870.
48. Shayeghi, M, Latunde-Dada GO, Oakhill JS, Laftah AH, Takeuchi K, Halliday N. Identification of an intestinal heme transporter. Cell 2005;122:789–801.
49. Qiu A, Jansen M, Sakaris A, Min SH, Chattopadhyay S, Tsai E, et al. Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell, 2006;127:917–28.
50. Gantz T, Nemeth E. Regulation of iron aquisition and iron distribution in mamals. Biochim Biophys Acta 2006;1763:690-9.
51. Ma Y, Yeh M, Yeh K, Glass J. Iron Imports V: Transport of iron through the intestinal epithelium. Am J Physiol Gastrointest Liver Physiol 2006;290:417-22.
52. Philpott CC. Coming into view: eukaryotic iron chaperones and intracellular iron delivery. J Biol Chem 2012;287:13518-23.
53. Shi H, Bencze KZ, Stemmler TL, Philpott CC. A cytosolic iron chaperone that delivers iron to ferritin. Science 2008;320:1207–10.
54. McKie AT, Marciani P, Rolfs A, Brennan K, Wehr K. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol Cell 2000;5:299-309.
55. McKie AT, Barlow DJ. The SLC40 basolateral iron transporter family (IREG1/ferroportin/MTP1). Pflugers Arch 2004;447:801-6.
56. Vulpe CD, Kuo YM, Murphy TL, Cowley L, Askwith C, Libina N, et al. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the SLA mouse. Nat Genet 1999;21:195-9.
57. Harris ZL, Durley AP, Man TK, Gitlin JD. Targeted gene disruption reveals an essential role for ceruloplasmin in cellular iron efflux. Proc Natl Acad Sci U S A 1999:96:10812-17.
58. Canonne-Hergaux F, Donovan A, Delaby C, Wang H, Gross P. Comparative studies of duodenal and macrophage ferroportin proteins. Am J Physiol Gastrointest Liver Physiol 2006;290:156-63.
59. Zhang LI, Senecal T, Ghosh MC, Ollivierre-Wilson H, Tu T, Roault TA. Hepcidin regulates ferroportin expression and intracellular iron homeostasis of erythroblasts Blood, 2011;118:2868-77.
60. Gkouvatsos K, Papanikolaou G, Pantopoulos K. Regulation of iron transport and the role of transferrin. Biochim Byophys Acta 2012;1820:188-202.
61. Baker HM, Anderson BF, Baker EN. Dealing with iron: Common structural principles in proteins that transport iron and heme. Proc Natl Acad Sci U S A 2003;100:3579-83.
62. Duk-Hee L, David R, Jacobs JR. Serum markers of stored body iron are not appropriate markers of health effect of iron: a focus on serum ferritin. Med Hypotheses 2004;62:442-5.
63. Brissot P, Ropert M, Lan CL, Loreal O. Non-transferrin bound iron: A key role in iron overload and iron toxicity. Biochim Biophys Acta 2012;1820:403-10.
64. Aisen P. Transferin receptor 1. Int J Biochem Cell Biol 2004;36: 2137-43.
65. Dassler K, Zydek M, Wandzik K, Kaup M, Fuchs H. Release of the soluble transferrin receptor is directly regulated by binding of its ligand ferritransferrin. J Biol Chem 2006;6:3297-304.
66. Gao J, Kramer J, Chen M, Tsukamoto H, Enns AS, Zhang CA. Interaction of the hereditary hemochromatosis protein HFE with transferin receptor 2 is required for transferrin-induced hepcidin expression. Cell Metab 2009;9:217-27.
67. Rapisarda C, Puppi J, Hughes RD, Dhawan A, Fernaud S, Evans RW, et al. Transferrin receptor 2 is crucial for iron sensing in human hepatocytes. Am J Physiol Gastrointest Liver Physiol 2010;299:G778-83.
68. Falzacappa MVV, Muckenthaler MU. Hepcidin: hormone and anti-microbial peptide. Gene 2005;364:37-44.
69. Goswami T, Andrews CN. Hereditary hemochromatosis protein, HFE: interaction with transferin receptor 1 suggests a molecular mechanism of iron sensing. J Biol Chem 2006;281:28494-8.
70. Wallace D, Summervile L, Subramaniam VN. Targeted disruption of hepatic transferrin receptor 2 gene in mice leads to iron overload. Gastroenterology 2007;132:301-10.
71. Ponka P, Lok CN. The transferrin receptor: role in health and disease. Biochem Cell Biol 1999;31:1111-37.
72. Ohgami RS, Campagna DR, Greer EL, McDonald B, Chen A, Antiochos J, et al. Identification of ferrireductase required for efficient transferrin-dependent iron uptake in erithroyd cells. Nat Genet 2005;37:1264-9.
73. Hider RC, Kong XL. Glutathione: a key component of the cytoplasmic labile iron pool. Biometals 2011;24:1179-87.
74. Andrews NC. Probing the iron pool. Focus on "Detection of intracellular iron by its regulatory effect". Am J Physiol Cell Physiol 2004;287:C1537-8.
75. Schneider BD, Leibold EA. Regulation of mammalian iron homeostasis. Curr Opin Clin Nutr Metab Care 2000;3:267-73.
76. Kakhlon O, Cabantchik ZI. The labile iron pool: characterization, and participation in cellular processes. Free Radic Biol Med 2002;8:1037-46.
77. Knutson MD, Oukka M, Koss LM, Aydemi F, Wessling-Resnick M. Iron release from macrophages after erythrophagocytosis is up-regulated by ferroportin1 overexpression and down-regulated by hepcidin. Proc Natl Acad Sci U S A 2005;102:1324-8.
78. Poss KD, Tonegawa S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc Natl Acad Sci U S A 1997;94:10919-24.
79. Roy C, Andrews NC. Anemia of inflammation: the hepcidin link. Curr Opin Hematol 2005;12:107-11.
80. Detivaud L, Nemeth E, Boudjema K, Turlih B, Troadec MB, Leroyer P, et al. Hepcidin levels in humans are correlated with hepatic iron stores, hemoglobin levels and hepatic function. Blood 2005;106:746-8.
81. Weinstein DA, Roy CN, Fleming MD, Loda MF, Wolfsdorf JI, Andrews C. Inappropriate expression of hepcidin is associated with iron refractory anemia: implication for anemia of chronic disease. Blood 2002;100:3776-81.
82. Veuthey T, D´Anna MC, Roque ME. Role of the kidney in iron homeostasis: renal expresion of prohepcidin, ferroportin, and DMT1 in anemic mice. Am J Physiol Renal Physiol 2008;295:F1213-21.
83. Kulaksiz, H Theiling F, Bachman S, Gherke SG, Rost D, Cetini A, Janetzko Y, et al. The iron regulatory protein hepcidin: expression and cellular localization in the mammalian kidney. J Endocrinol 2005;184:361-70.
84. Smith CP, Thevenod F. Iron transport and the kidney. Biochim Biophys Acta 2009;1790:724-30.
85. De Domenico I, Ward DM, Langelier C, Vaughn MB, Nemeth E, Sundquist WI, et al. The molecular mechanism of hepcidin-mediated ferroportin down-regulation. Mol Biol Cell 2007;18:2569-78.
86. Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004;306:2090-93.
87. Sangwaiya A, Manglam V, Busbridge M, Thursz M, Arnold J. Blunted increase in serum hepcidin as response to oral iron in HFE-hemochromatosis. Eur J Gastroenterol Hepatol 2011;23:721-4.
88. Anderson GJ, Frazer DM, McKie AT, Vulpe CD, Smith A. Mechanisms of haem and non-haem iron absorption: Lessons from inherited disorders of iron metabolism. Biometals 2006;18:339-48.
89. Chung AYF, Leo KW, Wong GC, Chuah KL, Ren JW, Lee CGL. Giant hepatocelullar adenoma presenting with chronic iron deficiency anemia. Am J Gastroenterol 2006;101:2160-2.
90. Nicolas G, Bennoun M, Porteu A, Mativet S, Beaumont C, Grandchamp B, et al. Severe iron deficiency anemia in transgenic mice expressing liver hepcidin. Proc Natl Acad Sci U S A 2002;99:4596-601.
91. Krause A, Neitz S, Magert HJ, Shulz A, Forssmann WG, Anderman P, Shulz-Knappe K. LEAP-1, a novel higly disulfide-bounded human peptide, exhibits antimicrobial activity. FEBS Lett 2000;750:147-50.
92. Park CH, Valore EV, Waring AJ, Ganz T. Hepcidin, a urinary antimicrobial peptide syntetized in liver. J Biol Chem 2001;276:7806-10.
93. Kulaksiz H, Gherke DG, Rost A, Janetzko D, Kallinowski T, Bruckner B, Stremmel W. Pro-hepcidin: expression and cell specific localisation in the liver and its regulation in hereditary haemochromatosis, chronic renall insufficiency, and renal anemia. Gut 2004;53:735-43.
94. Valore E, Ganz T. Posttranslational processing of hepcidin in human hepatocytes is mediated by the prohormon convertase furin. Blood Cells Mol Dis 2008;40:132-38.
95. Hunter HN, Fulton DB, Ganz T, Vogel HJ. The solution structure of human hepcidin, a peptide hormone with antimicrobial activity that is involved in iron uptake and hereditary hemochromatosis. J Biol Chem 2002;277:37597-603.
96. Gagliardo B, Kubat N, Faye A, Jaouen M, Durel B, Deschemin JC, et al. Pro-hepcidin is unable to degrade iron exporter ferroportin unless maturated by furin-dependent process. J Hepatol 2009;50:394-401.
97. Barthe C, Hocquellet A, Garbay B. Bacteriostatic activity of the proregion of human hepcidin. Protein Pept Lett 2011;18:36-40.
98. Zhang AS, Enns CA. Molecular mechanisms of normal iron homeostasis. Hematology Am Soc Hematol Educ Program 2009:207-14.
99. Kemna EH, Kartikasari AE, van Tits LJ, Pickkers P, Tjalsma H, Swinkels DW. Regulation of hepcidin: insights from biochemical analyses on human serum samples. Blood Cells Mol Dis 2008;40:339-46.
100. Drake SF, Herbison EH, Morgan CE, Delima R, Graham RM, Chua ACG, et al. Iron absorption and hepatic iron uptake are increased in transferrin receptor 2 (Y245X) mutant mouse model of hemochromatosis type 3. Am J Physiol Gastrointest Liver Physiol 2006;292:G323-8.
101. Niederkofler V, Salie R, Arber S. Hemojuvelin is essentioal for dietary iron sensing, and its mutation leads to severe iron overload. J Clin Invest 2005;115:2180-6.
102. Merle U, Theiling F, Fein E, Gherke S, Riedel B, Kallinowski HD, et al. Localization of the iron-regulatory proteins hemojuvelin and transferrin receptor 2 to the basolateral domain of hepatochytes. Histochem Cell Biol 2007;127:221-6.
103. Chen J, Enns C. Hereditary hemochromatosis and transferrin receptor 2. Biochim Biophys Acta 2012;1820:256-63.
104. Papanikolaou G, Samuels ME, Ludwig EH, MacDonald MLE, Franchini PL, Dube MP, et al. Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat Genet 2004;36:77-82.
105. Babitt JL, Huang WH, Wrighting DM, Xia Y, Sidis Y, Samad TA, et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat Genet 2006;38:531-39.
106. Bartnikas TB, Fleming MD. Hemojuvelin is essential for transferrin-dependent and transferrin-independent hepcidin expression in mice. Haematologica 2012;97:189-92.
107. Du X, She E, Gelbart T, Truksa J, Lee P, Xia Y, et al. The serine protease TMPRSS6 Is required to sense iron deficiency. Science 2008;320:1088-92.
108. Finberg KE, Whittlesey RL, Fleming MD, Andrews NC. Down-regulation of Bmp/Smad signaling by Tmprss6 is required for maintanance of systemic iron homeostasis. Blood 2010;115:3817-26.
109. Krijt J, Fujikura Y, Ramsay AJ, Velasco G, Nečas E. Liver hemojuvelin protein levels in mice deficient in matriptase-2 (Tmprss6). Blood Cells Mol Dis 2011;47:133-7.
110. Lee P, Peng H, Gelbart T, Beutler E. The IL-6 and lipopolysaccharide-induced transcription of hepcidin in HFE, transferrin receptor 2 and beta-2-microglobulin-deficient hepatocytes. Proc Natl Acad Sci U S A 2004;101:9263-5.
111. Nemeth E, Valore EV, Territo M, Schiller G, Lichenstein A, Ganz T. Hepcidin, putative mediator of anemia of inflammation, is type II acute-phase protein. Blood 2003;101:2461-3.
112. Kemna EP, Nemeth E, van der Hoeven H, Pickkers, Swinkels D. Time course analysis of hepcidin, serum iron, and plasma cytokine levels in humans injected with LPS. Blood 2005;106:1864-6.
113. Wrighting D, Andrews NC. Interleukin-6 induces hepcidin expression through STAT3. Blood 2006;108:3204-9.
114. Nemeth E, Rivera S, Gabayan V, Keller C, Taudorf S, Pedersen BK, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest 2004;113:1271-6.
115. Khan FA, Fisher MA, Khakoo RA. Association of hemochromatosis with infectious diseases: expanding spectrum. Int J Infect Dis 2007;11:482-7.
116. Ong ST, Ho JZ, Ho B, Ding JL. Iron-withholding strategy in innate immunity. Immunobiology. 2006;211:295-314.
117. Theur I, Schroll A, Nairz M, Selfert M, Theurl M, Sonnweber T, et al. Pathways for the iron regulation of hepcidin expression in anemia of chronic disease and iron deficiency anemia in vivo. Haematologica 2011;96:1761-9.
118. Vyoral D, Petrak J. Hepcidin: A direct link between iron metabolism and immunity Int J Biochem Cell Biol 2005;37:1768-73.
119. Nicolas G, Chauvet C, Viatte L, Danan JL, Bigard X, Devaux I, et al. The gene encoding iron regulatory peptide hepcidin is regulated by anemia, hypoxia and inflammation. J Clin Invest 2002;110:1037-44.
120. Pak M, Gabayan MA, Lopez V, Ganz T, Rivera S. Suppression of hepcidin during anemia requires erythropoietic activity. Blood 2006;12:3730-5.
121. Vokurka M, Krijt J, Sulc K, Necas E. Hepcidin mRNA levels in mouse liver respond to inhibition of erythropoiesis. Physiol Res 2006;55:667-74.
122. Kattamis A, Papassotiriou I, Palaiogou D, Galani F, Apostolakou A, Ladis V, et al. The effect of erythropoetic activity and iron burden on hepcidin expression in patients with thalassemia major. Haematologica 2006;91:809-12.
123. Pinto JP, Ribeiro S, Pontes H, Thowfeequ S, Tosh D, Carvalho F, et al. Erythropoietin mediates hepcidin expression in hepatocytes through EPOR signaling and regulation of C/EBPalpha. Blood 2008;111:5727–33.
124. Tanno T, Bhanu NV, Oneal PA, Goh SH, Staker P, Lee YT, et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat Med 2007;13:1096–101.
125. Tanno T, Porayette A, Orapan S, Noh SJ, Byrnes C, Bhupatiraju A, et al. Identification of TWSG1 as a second novel erythroid regulator of hepcidin expression in murine and human cells. Blood 2009;114:181–6.
126. Lakhal S, Schödel J, Townsend ARM, Pugh CW, Ratcliffe PJ, Mole DR, et al. Regulation of type II transmembrane serine proteinase TMPRSS6 by hypoxia-inducible factors. J Biol Chem 2011;286:4090-7.
127. Peyssonnaux C, Zinkernagel AS, Schuepbach RA, Rankin E, Vaulont S, Haase VH, et al. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). J Clin Invest 2007;117:1926–32.
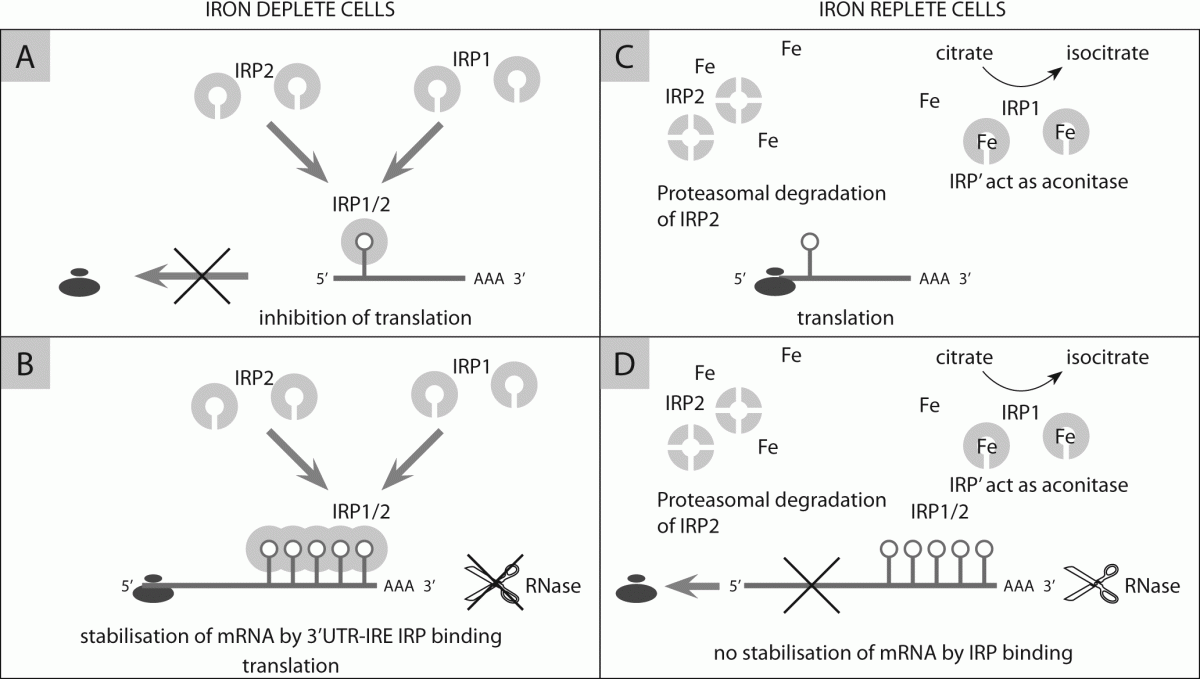
128. Sanchez M, Galy B, Schwanhaeusser B, Blake J, Bähr-Ivacevic T, Benes V, et al. Iron regulatory protein-1 and -2: transcriptome-wide definition of binding mRNAs and shaping of the cellular proteome by iron regulatory proteins. Blood 2011;118:168-79.
129. Beaumont C, Leneuve P, Devaux I, Scoazec JY, Berthier M, Loiseau MN, et al. Mutation in the iron responsive element of the L ferritin mRNA in a family with dominant hyperferritinaemia and cataract. Nat Genet 1995;11:444-6.
130. Kato J, Fujikawa K, Kanda M, Fukuda N, Sasaki K, Takayama T, et al. A mutation, in the iron-responsive element of H ferritin mRNA, causing autosomal dominant iron overload. Am J Hum Genet 2001;69:191-7.
131. Clarke SL, Vasanthakumar A, Anderson SA, Pondarre C, Koh CM, Deck KM, et al. Iron-responsive degradation of regulatory protein 1 does not require the Fe-S cluster. EMBO J 2006;25:544-53.
132. Meyron-Holtz EG, Gosh MC, LaVaute I, Brazzolotto T, Kazuhiro X, Berger UV, et al. Genetic ablations of iron regulatory proteins 1 and 2 reveal why iron regulatory protein 2 dominates iron homeostasis. EMBO J 2004;23:386-95.
133. Eisenstein RS, Biemings KP. Iron regulatory proteins, iron responsive elements and iron homeostasis. J Nutr 1998;128:2295-98.
134. Sasu BJ, Cooke KS, Arvedson TI, Plewa C, Ellison AR, Sheng J, et al. Antihepcidin antibody treatment modulates iron metabolism and is effective in a mouse model of inflammation-induced anemia. Blood 2010;115:3616-24.
135. Gantz T. Hepcidin in iron metabolism. Curr Opin Hematol 2004;11:251-4.
136. Pinnix ZK, Miller LD, Wang W, D`Agostino R, Kute T, Williongham MC, et al. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci Transl Med 2010;43:43-5.
137. Chaston TB, Wats RN, Yuan J, Richardson DR. Potent antitumor activity of novel iron chelators derivied from di-2-pyridylketone isonicotinyl hydrazone involves fenton-derived free radical generation. Clin Cancer Res 2004;10:7365-74.
138. Chantrel-Groussard K, Gaboriau F, Pasdeloup N, Havouis R, Nick H, Pierre JL, et al. A new orally active iron chelator ICL670A exhibits a higher antiproliferative effect in human hepatocyte cultures than O-trensox. Eur J Pharmacol 2006;541:129-37.
139. Whitnall M, Howard J, Ponka P, Ritchardson DR. A class of iron chelators with a wide spectrum of potent antitumor activity that overcomes resistance to chemotherapeutics. Proc Natl Acad Sci U S A 2006;103:14901-6.
140. Quian ZM, Li H, Sun H, Ho K. Targeted drug delivery via the transferrin-receptor mediated endocytosis pathway. Pharmacol Rev 2002;54:561-84.
141. Li H, Sun H, Quian ZM. The role of the transferrin-transferrin receptor- system in drug delivery and targeting. Trends Pharmacol Sci 2002;23:206-9.
142. Li X, Jankovic L, Lee W. Iron chelation and neuroprotection in neurodegenerative diseases. J Neural Transm 2011;118:373-7.
143. Avramovich-Tirosh Y, Amit T, Bar-Am O, Zheng H, Fridkin M, Youdim MBH. Therapeutic targets and potential of the novel brain-permeable multifunctional iron chelator-monoamine oxidase inhibitor drug, M-30, for the treatment of Alzheimer´s disease. J Neurochem 2007;100:490-502.
144. Kelly R, Giaccone G. The role of talactoferrin alfa in the treatment of non-small cell lung cancer. Expert Opin Biol Ther 2010;10:1379-86.
145. Weinberg, ED. Human lactoferrin: a novel therapeutic with broad spectrum potential. J Pharm Pharmacol 2001;53:1303-10.
146. Weinberg, ED. Iron withholding: a defense against viral infections. Biometals 1996;9:393-9.
147. Berlutti F, Pantanella F, Natalizi T, Frioni A, Paesano R, Polimeni A, et al. Antiviral properties of lactoferrin: A natural immunity molecule. Molecules 2011;16:6992-7018.
148. Ganz T, Olbina G, Girelli D, Nemeth E, Westerman M. Immunoassay for human serum hepcidin. Blood 2008;112:4292-7.
149. White KN, Conesa C, Sánchez L, Amini M, Farnaud S, Lorvoralak C, et al. The transfer of iron between ceruloplasmin and transferrins. Biochim Biophys Acta 2012;1820:411-6.
150. Ward PP, Conneely OM. Lactoferrin: role in iron homeostasis and host defense against microbial infection. Biometals 2004:17:203-8.
151. Bao G, Clifton M, Hoette TM, Mori K, Deng SX, Qiu A et al. Iron traffics in circulation bound to a siderocalin (Ngal)-catechol complex. Nat Chem Biol 2010;6:602-9.
152. Pandolfo M, Pastore A. The pathogenesis of Friedreich ataxia and the structure and function of frataxin. J Neurol 2009;256(Suppl. 1):9-17.
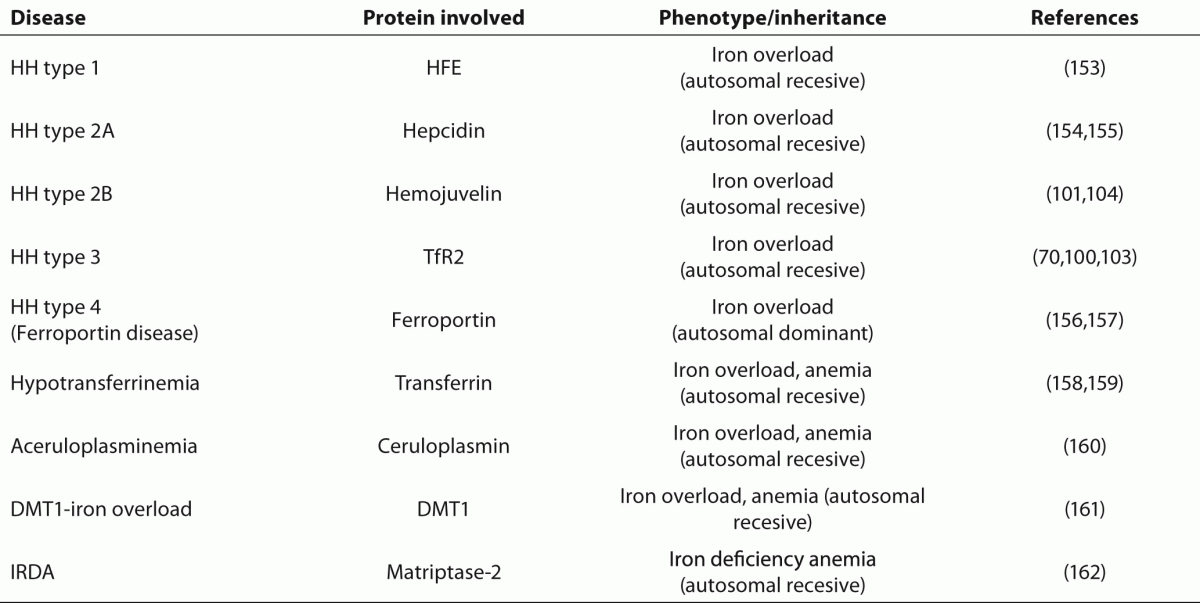
153. FederJN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet 1996;13:399-408.
154. Hattori A, Tomosugi N, Tatsumi Y, Suzuki A, Hayashi K, Katano Y, et al. Identification of a novel mutation in the HAMP gene that causes non-detectable hepcidin molecules in a Japanese male patient with juvenile hemochromatosis. Blood Cells Mol Dis 2012;48:179-82.
155. Roetto A, Papanikolaou G, Politou M, Alberti F, Girelli D, Christakis J, et al. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis Nature Genetics 2002;33:21-2.
156. Njajou OT, Vaessen N, Joosse M, Berghuis B, van Dongen JW, Breuning MH, et al. A mutation in SLC11A3 is associated with autosomal dominant hemochromatosis.
Nat Genet 2001;28:213-4.
157. Montosi G, Donovan A, Totaro A, Garuti C, Pignatti E, Cassanelli S, et al. Autosomal-dominant hemochromatosis is associated with a mutation in the ferroportin (SLC11A3) gene.
J Clin Invest 2001;108:619-23.
158. Hamill RL, Woods JC, Cook BA. Congenital atransferrinemia. A case report and review of the literature.
Am J Clin Pathol1991;96:215-8.
160. Yoshida K, Furihata K, Takeda S, Nakamura A, Yamamoto K, Morita H et al. A mutation in the ceruloplasmin gene is associated with systemic hemosiderosis in humans Nature Genetics 1995;9:267-72.
161. Mims MP, Guan Y, Pospisilova D, Priwitzerova M, Indrak K, Ponka P, et al. Identification of a human mutation of DMT1 in a patient with microcytic anemia and iron overload. Blood 2005;105:1337-42
162. De Falco L, Totaro F, Nai A, Pagani A, Girelli D, Silvestri L, et al. Novel TMPRSS6 mutations associated with iron-refractory iron deficiency anemia (IRIDA).
Hum Mutat. 2010;31:E1390-405.