Introduction
Congenital analbuminemia (CAA) (OMIM # 103600) is an autosomal recessive disorder manifested by the presence of a very low amount of circulating serum albumin, in the absence of hepatic dysfunction, renal or gastrointestinal losses, and redistribution into extra-vascular compartments (1,2). In the affected individuals albumin concentration may range from < 1 g/L up to 10 g/L (2). This wide interval is probably due to the fact that the commonly used assays for the quantification of the protein in serum (based on dye-binding techniques) suffer from poor accuracy in presence of low levels, usually showing an overestimation of its real amount (3).
CAA is very rare: in spite of the fact that the trait is readily detected by routine plasma electrophoretic analysis, only just over fifty cases have been so far reported world-wide and are listed in the Register of Analbuminemia Cases (4). Its prevalence is estimated at less than 1 in 1 million, apparently without gender or ethnic predilection (2). Recent data, however, seem to indicate that the frequency of the trait is significantly higher in restricted and minimally admixed population groups, such as a gypsy settlement in Slovakia and two Amerindian communities in Saskatchewan (Canada), than in the average population (2).
In the adult population, CAA is generally thought as a benign condition, since the absence of the major blood protein is partially compensated for by an increase in the synthesis of other serum proteins (2,5,6). As reported in the Register of Analbuminemia Cases, analbuminemic individuals have few clinical symptoms of their condition other than mild edema, hypotension, fatigue and, occasionally, a peculiar lower-body lipodystrophy (mainly in adult females), while the most common biochemical finding is a gross hyperlipidaemia, with a significant increase in the total and LDL-cholesterol concentration, but normal concentration of HDL-cholesterol and triglycerides. Due to the lack of follow-up data in most cases, it is hard to draw conclusions about the long-term outcome, and the possibility that analbuminemic individuals may be at risk for atherosclerotic complications cannot be ruled out (2).
In contrast to the benign presentation of CAA after birth, the prenatal course appears less favourable. Fetal or neonatal death of siblings was frequently observed in the families of analbuminemic subjects, suggesting that albumin has a crucial role in fetal development (2,5,6). A recent study shows that CAA is a risk factor during the perinatal and the childhood period (7), confirming the hypothesis that the rarity of the trait may be attributed to the fact that only a few analbuminemic individuals survive past the neonatal state (8,9).
All the cases of CAA so far studied at the molecular level are caused by mutations in the albumin gene (ALB), which lies on chromosome 4, near the centromere, at position 4q11-13 (74269972..74287129) (10). In a continuing effort towards understanding the molecular basis of CAA, we report here the clinical findings and the mutational analysis of the ALB in a new case of analbuminemia diagnosed in a Turkish female new-born.
Materials and methods
Subjects and clinical laboratory analyses
Subjects of this study were the members of a family, composed by consanguineous (first degree cousins) healthy parents from Ankara, Turkey, and by a female new-born (the index case). At the time of our study the mother was 19 years old and the father was 26 years old. The albumin concentration was measured also in 12 healthy relatives, aged between 14-60 years. Those samples could not be submitted to genetic analysis. The clinical and biochemical studies were performed at the Department of Pediatrics, Division of Neonatology, of the Ankara University School of Medicine, Ankara, Turkey in November 2012. The mutational analysis was done in 2013 at the Department of Molecular Medicine, University of Pavia, Pavia, Italy and at Laboratory on Pathophysiology of Uremia, Istituto Giannina Gaslini IRCCS, Genova, Italy.
The procedures were in accordance with the 1975 Helsinki Declaration. We collected blood samples after obtaining written informed consent from all participants involved in the study. Albumin concentration in serum was measured by a modified bromcresol purple-binding assay. Serum protein electrophoresis was performed by the fully automated capillary electrophoresis system Capillarys 2® (Sebia, Norcross, GA, USA), designed to optimise and completely automate electrophoresis testing. The method uses an alkaline buffer (Protein 6 Assay, Sebia) for a high voltage protein separation. Proteins are detected by measuring UV absorbance of the peptide bonds at 214 nm at the cathodic capillary end. The system automatically generates a profile that resembles patterns obtained after conventional gel electrophoresis on cellulose acetate or agarose. Other analytes were assayed by conventional techniques using a fully automated Beckman Coulter, DXC 800 Autoanlyser (Beckman Coulter Inc., Brea, CA, United States).
Mutational analysis
Fourteen genomic fragments of the ALB gene, encompassing the coding exons and their intron-exon junctions (10), were amplified by PCR using specific primer pairs as described by Watkins et al. (9). Genomic DNA from two unrelated volunteers was available as a control. Heteroduplex and single strand conformational polymorphism analysis and DNA sequencing were performed as previously reported (11,12).
White blood cells, separated from whole blood by using Ficoll solution, were used for isolation of total RNA with the RNeasy Mini Kit (Qiagen, Venlo, Netherlands) from the proband and from one control. 1 μg of extracted RNA was used in reverse-transcription reactions by using 10 pmol of oligo (dT) primers and the Quantitec reverse transcription enzyme (Qiagen). The first strand cDNA was amplified by primers specific for exon 9 (ALB_1117F: 5’-CTT GCC AAG ACA TAT GAA ACC A-3’) and exon 14 (ALB_1816R (5’-CAG CTT GAC TTG CAG CAA CA-3’). The PCR products obtained were subjected to direct sequencing as described above.
As NCBI reference sequences we used NG_009291.1 for ALB, NM_000477.5 for the messenger RNA, and NP_000468.1 for the precursor protein (preproalbumin) of 609 amino acid residues.
Results
Patient
The proband was bornat 333/7 gestational week from the second pregnancy, which was complicated by hyperemesis gravidarum up to third trimester and intrauterine growth retardation (IUGR). The mother’s first pregnancy resulted with abortion at 8th gestational week. The new-born was delivered by caesarean section due to non-reactive stress test on follow-up for IUGR. She was small for gestational age with birth weight of 1130 g (< 3rd percentile) and length of 39 cm (< 10th percentile). She was admitted to neonatal intensive care unit for respiratory distress observed in delivery room in addition to IUGR. Clinical examination was normal except for respiratory insufficiency which was managed by nasal continuous positive airway pressure for only four hours. Complete blood count on admission revealed polycythaemia (haematocrit 0.670), and partial exchange transfusion was performed due to concurrently hypoglycaemia. Albumin concentration in serum, measured by a modified bromcresol purple-binding assay at 24th h of life resulted abnormally low (< 8 g/L). This test was repeated for three times, giving the same result. Automated capillary electrophoresis of serum proteins confirmed the presence of a similarly low albumin concentration associated with a compensatory increase in the non-albumin protein, especially the α2- and the β-globulin fractions (Figure 1 and Table 1). Other relevant clinical chemistry data of the proband are reported in Table 1. Besides the above mentioned severe hypoalbuminemia and moderate hypoproteinemia, she exhibits moderate dyslipidemia, with a marked increase of the triglyceride concentration, whereas the concentration of total cholesterol, LDL-cholesterol and HDL-cholesterol are close to the upper limit of the normal range. In addition, the proband shows moderate hypocalcemia, related to her low albumin concentration (1,13).The clinical condition of the patient deteriorated on postnatal third day, and she developed pulmonary haemorrhage, upper gastrointestinal system bleeding and haematuria. She was diagnosed as disseminated intravascular coagulation related to sepsis. She received antibiotics (vancomycin and amikacin), and supported with multiple fresh frozen plasma while she was on respiratory support with surfactant and high frequency ventilation. Patient received albumin infusion as 1 g/kg, and the protein concentration increased to 17 g/L. On postnatal 6th day, she was extubated and her clinical condition was stable up to 24th day when she had minimal edema on dorsum of hands and feet. The albumin concentration was again low, and she received one additional infusion for abdominal distension, which was thought to be related to intestinal edema.
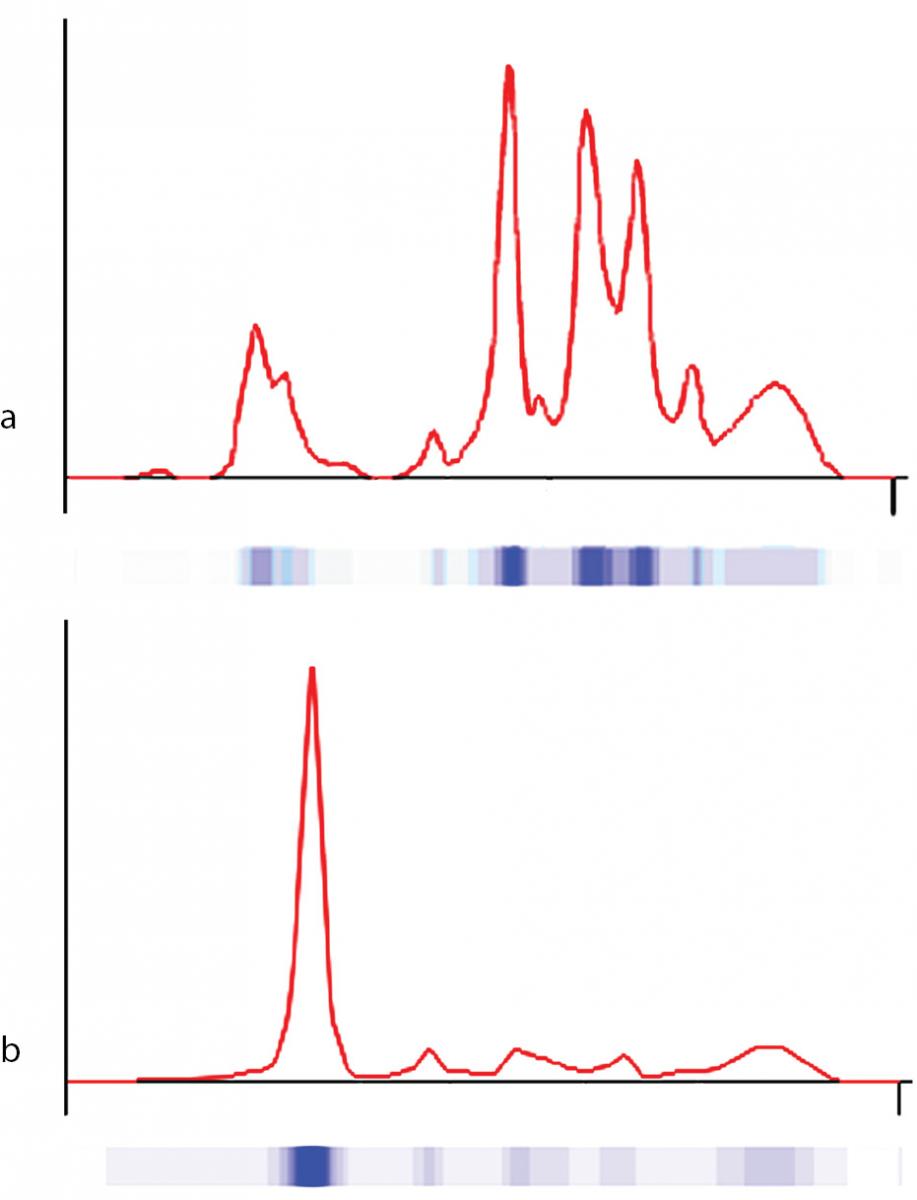
Figure 1. Capillary electrophoresis of serum proteins. The profiles were obtained via the fully automated capillary electrophoresis system Capillarys 2® as described in Material and methods. a) patient (on postnatal second day); b) control.
Table 1. Clinical laboratory test results of the analbuminemic patient.
Differential diagnosis led us to congenital nephrotic syndrome and congenital analbuminemia because of normal gastrointestinal system findings and age of the patient. Renal ultrasonography revealed normal parenchymal echogenity with normal size. 24 h collected urine analysis showed proteinuria (15-20.5 mg/m2/h, normal value < 4 mg/m2/h) which was never above nephrotic level (> 40 mg/m2/h). Proteinuria resolved on follow-up (2.6 mg/m2/h) in the neonatal intensive care unit, and was thought to be related with sepsis and haematuria. On the basis of the above-reported clinical and biochemical findings, CAA was diagnosed. A relatively low albumin concentration was detected in the mother (24 g/L), in the father (36 g/L) and in five other members of the family (Figure 2). This suggests that all of them are heterozygous carriers of a mutation, which, at the homozygous or compound heterozygous state, may result in CAA, since those individuals generally have albumin concentrations close to the lower limit of the normal range (about 30-35 g/L), an otherwise unremarkable condition (1).
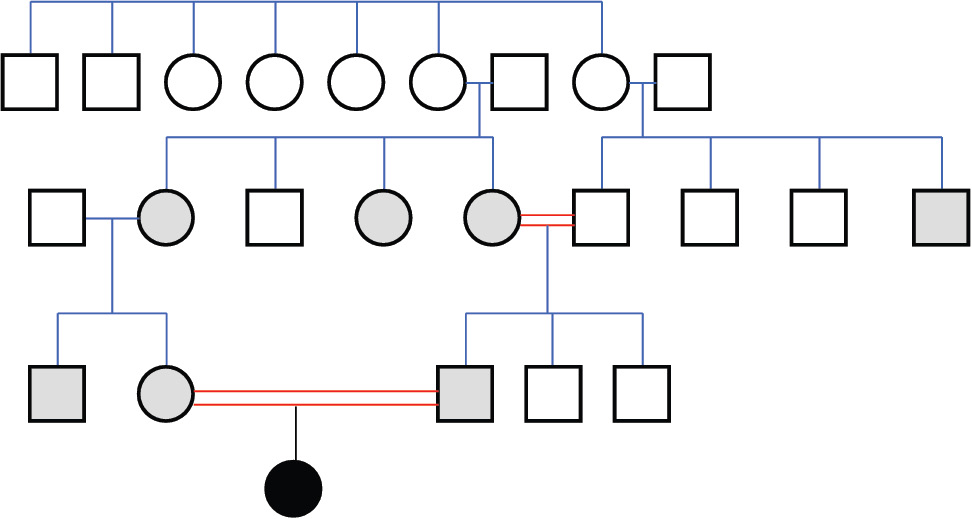
Figure 2. Family pedigree.
The analbuminemic patient is represented by a black symbol. The albumin levels were determined in all the members of the second and third generation. Grey symbols indicate individuals who have albumin concentrations close to the lower limit of the normal range (about 30-35 g/L) and are therefore suspected to be heterozygous for the mutation. Heterozygosity was demonstrated for the parents of the proband.
On out-patient follow-up, the patient’s physical examination was completely normal with a weight of 7890 g (3rd-10th percentile) at one year of corrected age. The patient’s all biochemical evaluation on six out-patient follow-up during the first year of life revealed albumin concentration below 8 g/L without any sign or symptom.
Mutational analysis
To confirm the diagnosis of CAA at the molecular level, a mutational analysis of the ALB was carried out as described in Materials and methods. In this case the combination of heteroduplex analysis and single-strand conformation polymorphism did not allow an unambiguous localisation of the mutation site. Therefore all the fourteen coding exons and their adjacent intron regions were submitted to DNA sequence analysis, which clearly indicated the presence of a single mutation in the region encompassing exon 12 and the intron 11 - exon 12 and exon 12 - intron 12 junctions, that escaped our electrophoretic analysis. The electropherogram of the proband revealed that she is homozygous for a G>A transition at position c.1652+1, the first base of intron 12 in a 5’ GT consensus sequence (Figure 3a). Both parents were heterozygous for the wild-type and mutated alleles, as the sequencing electropherograms showed the presence of two superimposed peaks at nucleotide c.1652+1 (Figure 3b and 3c). To establish the consequences of the present splicing mutation, we attempted to amplify ALB cDNA from the proband and from a wild type control. Amplified products showed different sizes: a PCR band of 476 bp was amplified from the proband cDNA, whereas a 700 bp fragment was obtained from the control (data not shown). Sequencing of the patient PCR product allowed us to establish that the G>A transition at the first position of intron 12 results in the complete skipping of the preceding exon (Figure 3a and 3b). The subsequent frame-shift within exon 13 causes the translation of three mutant amino acid residues (Cys-Thr-Cys) and a premature stop codon (TGA) located at amino acid position 480 (Figure 3c). The putative protein product, according to the Human Genome Variation Society rules, is p.Leu477Cysfs4*, indicating a frame-shifting change with Cysteine 477 as the first affected amino acid and the new reading frame being open for three amino acids (Figure 3c). This extensive modification of the C-terminal region of ALB should give rise to a putative polypeptide chain of 455 amino acid residues, with a relative molecular mass of 51,651 and a theoretical pI of 5.61, instead of 66,472 and 5.67 of the normal molecule. Unfortunately, because of the very young age of the patient and of her not good clinical condition, we did not have the possibility to perform two-dimensional electrophoresis, in order to check the presence in serum of this truncated albumin molecule. These results show that the splicing mutation here described, for which we suggest the name Ankara after the city of origin of the family, affects pre-mRNA maturation by inactivating the 5’ splice site sequence at the twelfth exon-intron boundary of the ALB, and represents a novel genetic defect causing CAA.
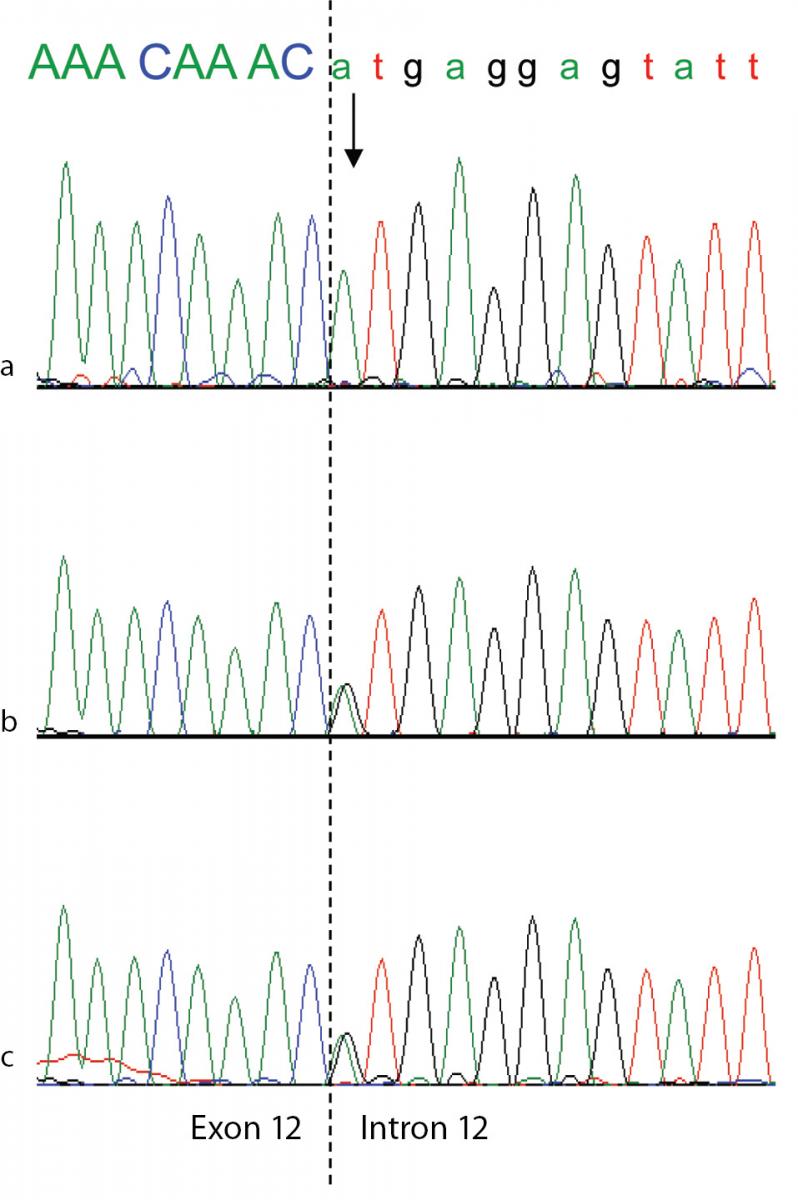
Figure 3. Genomic DNA sequence electropherograms of the analbuminemic patient and her parents.
The arrow indicates the nucleotide (A) which in this family substitutes the G at position c.1652 +1, the first base of intron 12 in a 5’ GT consensus sequence. The patient (a) is homozygous for the mutation, while both parents (b and c) are heterozygous for the wild-type and mutated alleles, as seen by the presence of two superimposed peaks.
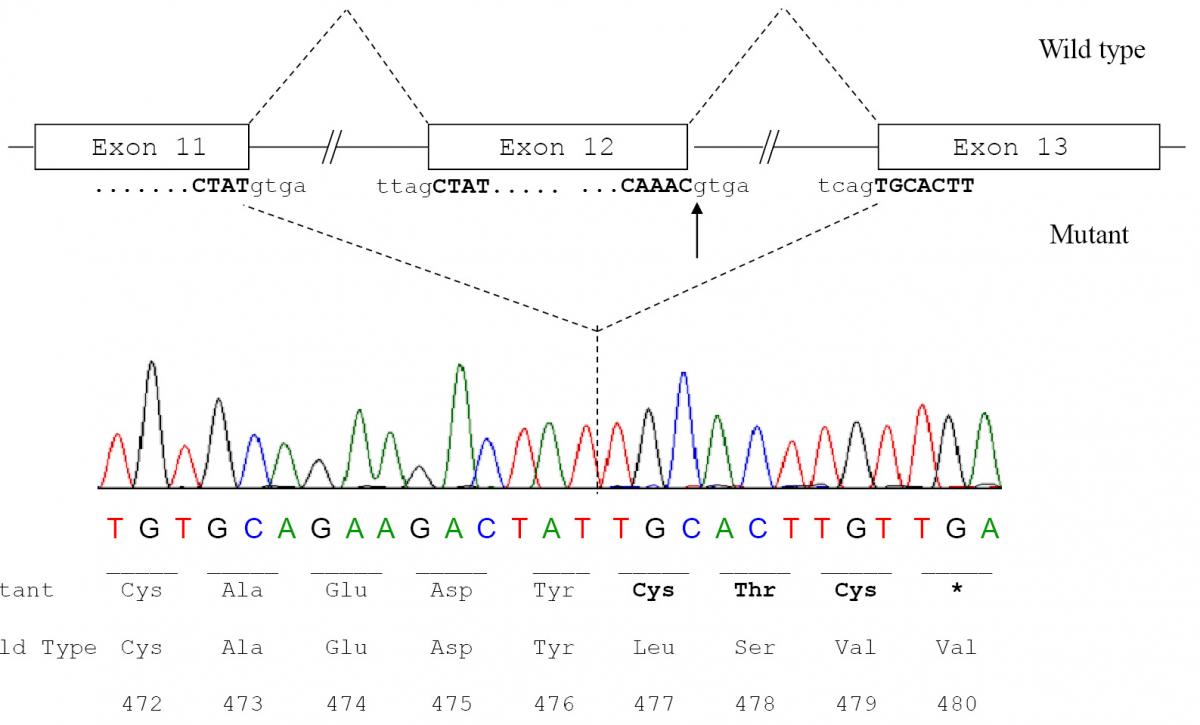
Figure 4. Molecular effect of the novel c.1652+1G>A mutation.
a) Schematic representation of normal and aberrant mRNA splicing in the ALB. The arrow indicates the nucleotide (the G at the first position of intron 12) which is mutated in the patient; b) ALB cDNA sequence from the patient showing that the G>A transition at position c.1652+1, which causes the inactivation of the strongly conserved GT dinucleotide at the 5’ splice site consensus sequence of intron 12, results in the splicing between exons 11 and 13 with the complete skipping of exon 12; c) Predicted amino acid sequence of the C-terminal region of the putative variant protein product. The subsequent frame-shift within exon 13 causes the translation of three mutant amino acid residues (Cys-Thr-Cys) and a premature stop codon (TGA) located at amino acid position 480. The variant amino acids and the stop codon introduced as a consequence of the mutation are in bold.
Discussion
Direct DNA sequencing of the ALB in the index case allowed the identification of a novel splicing defect. The subsequent aberrant splicing could be verified at the cDNA level. The results confirmed the clinical diagnosis of CAA in the proband and the inheritance of the trait from the two heterozygous parents. The only limitation of this study was the impossibility of testing the presence of the mutation in other members of the family. Pre-term births, presence of miscarriages in the history of the affected families, and respiratory tract infections with frequent hospital admissions have been reported in analbuminemic new-borns, who are often small for their gestational age (7). All those signs, together with other clinical complications, were present in the index case. This seems to confirm that albumin has a crucial role in the fetal development and its absence is a risk factor in the perinatal and in childhood period (2,7-9).
The first reported cases of CAA were diagnosed in the adult age (4). In the following years, more attention to this condition and a more detailed knowledge of the trait led to an earlier diagnosis, often in the paediatric age (4). The analbuminemic individuals, however, are prone to misdiagnosis or delayed diagnosis, because of the absence of unambiguous symptoms and of the presence of a significant, though low, amount of albumin. The major pitfall is that a minute albumin amount, as reported in the Introduction, may have many causes different from a genetic lack of the protein. For these reasons, the clinical diagnosis needs always to be confirmed by a molecular diagnosis, based on the identification of the causative mutation within the ALB.The Ankara mutation is the twenty-second different molecular defect identified as a cause of the analbuminemic trait (2,11). Twenty among them cause CAA at the homozygous state: six nonsense mutations, seven mutations affecting splicing, five frame-shift/deletions, one frame-shift/insertion and one mutation in the start codon (2,11). Compound heterozygosity for the remaining two molecular defects, a nonsense mutation and a splice site mutation with subsequent reading frame-shift, caused CAA in an Italian man (2,11).
The length of the truncated aberrant albumins, produced as a consequence of these mutations, would range from 31 to 532 amino acid residues, but, to date, no evidence could be found for the presence of these putative protein products in the serum of analbuminemic individuals (2). A possible explanation for their absence is that all these truncated proteins will partly or completely lack domain III. The C-terminal end of the molecule is crucial for binding to the intracellular receptor FcRn, which acts to recycle the protein back to the blood (14). Alternatively, since all the mutations associated with CAA occur at least 55 bp upstream of the exon13-intron13 boundary, all truncated transcripts could undergo nonsense-mediated RNA decay with no protein produced (15).
The exon/intron junctions in the ALB conform with the invariant dinucleotides GT and AG consensus sequences present at the 5’(donor) and 3’(acceptor) splice sites, respectively (10,16). In humans, mutations that affect pre-mRNA splicing may have a dramatic impact on the structure, or outcome, of mature transcripts, on the function of their translation products and may cause phenotypic manifestations (17). The most common consequence of splicing mutations is skipping of one or more exons, followed by the activation of aberrant 5’ donor or acceptor splice sites and retention of full introns in mRNA (17). Among the eight splice-site mutations, five affect the GT consensus dinucleotide sequence at the donor intron splice site: Baghdad (c.79+1G>A) (18), Bartin (c.1428+2T>C) (19), Tripoli (c.1428+1G>T) (20), Guimarães (c.1289+1G>A) (21), and Ankara (present paper). Although the presence of truncated albumin molecules in the serum could never be evidenced in analbuminemic individuals, in all the four cases (Fondi, Bartin, Guimarães and Ankara) in which there was performed a search for mRNA, it could be isolated from white blood cells, which allowed for a verification of the splicing defects at the mRNA level (2 and present paper). Thus, these four splicing mutations did not cause a complete degradation of the variant mRNA. As in the present case, in analbuminemia Bartin and Guimarães the mutations resulted in the skipping of the preceding exon (19,21), whereas in analbuminemia Baghdad and Tripoli the consequence of the mutation on mRNA could not be evaluated (18,20).
The Ankara mutation, as the vast majority of the defects which cause CAA, is so far unique, having been found in a single individual, or in members of the same family or closely related families (2). Only in two cases, the same mutation was found in two unrelated subjects (2). In contrast, the two bases deletion c.228_229del (analbuminemia Kayseri) has been ascertained in thirteen analbuminemic individuals of nine families belonging to geographically distant and apparently distinct ethnic groups (2). In addition, it is very likely the cause of CAA in about twenty other subjects (2,7). Therefore, it is by far the most frequent cause of the trait so far identified, accounting for about 40% of the cases characterised at the molecular level and, probably, for about half of the known affected individuals world-wide (2).
The twenty-two molecular defects known to result in CAA are located in nine different exons (1, 3, 4, 5, 7, 8, 10, 11, and 12) and in five different introns (1, 6, 10, 11, and 12). This pattern seems to suggest that CAA is the result of widely scattered random mutations (22). However, as the number of the known molecular defects is increasing, the data seem to point out to the presence of regions in the ALB that are prone to mutations resulting in CAA. Two of them appear to be localised in the intron 6-exon 7 and exon 11-intron 11 junctions (2, 11). Other hypermutable regions seem to be the sequence c.228 – c.230 of exon 3, the CpG sequence at position c.412 – 413 in the codon CGA for Arg138 in exon 4, and the sequence c.1610 – 1615 near the 3’ end of exon 12 (2,11).