Introduction
Parallel with better understanding of biochemical processes in bones and isolation and characterization of cellular components of skeletal matrix, the number of new potential biochemical markers of bone formation and resorption is increasing. Generally, markers are classified into the following groups:
a) enzyme activity markers of bone formation (connected with osteoblast activity) and of bone resorption (connected with osteoclast activity);
b) bone matrix proteins and resorption products of organic skeletal matrix, which are released into circulation during bone formation and resorption; and
c) inorganic skeletal matrix markers (calcium, phosphorus which, above all, reflect calcium-phosphorus homeostasis and which will not be considered in this review).
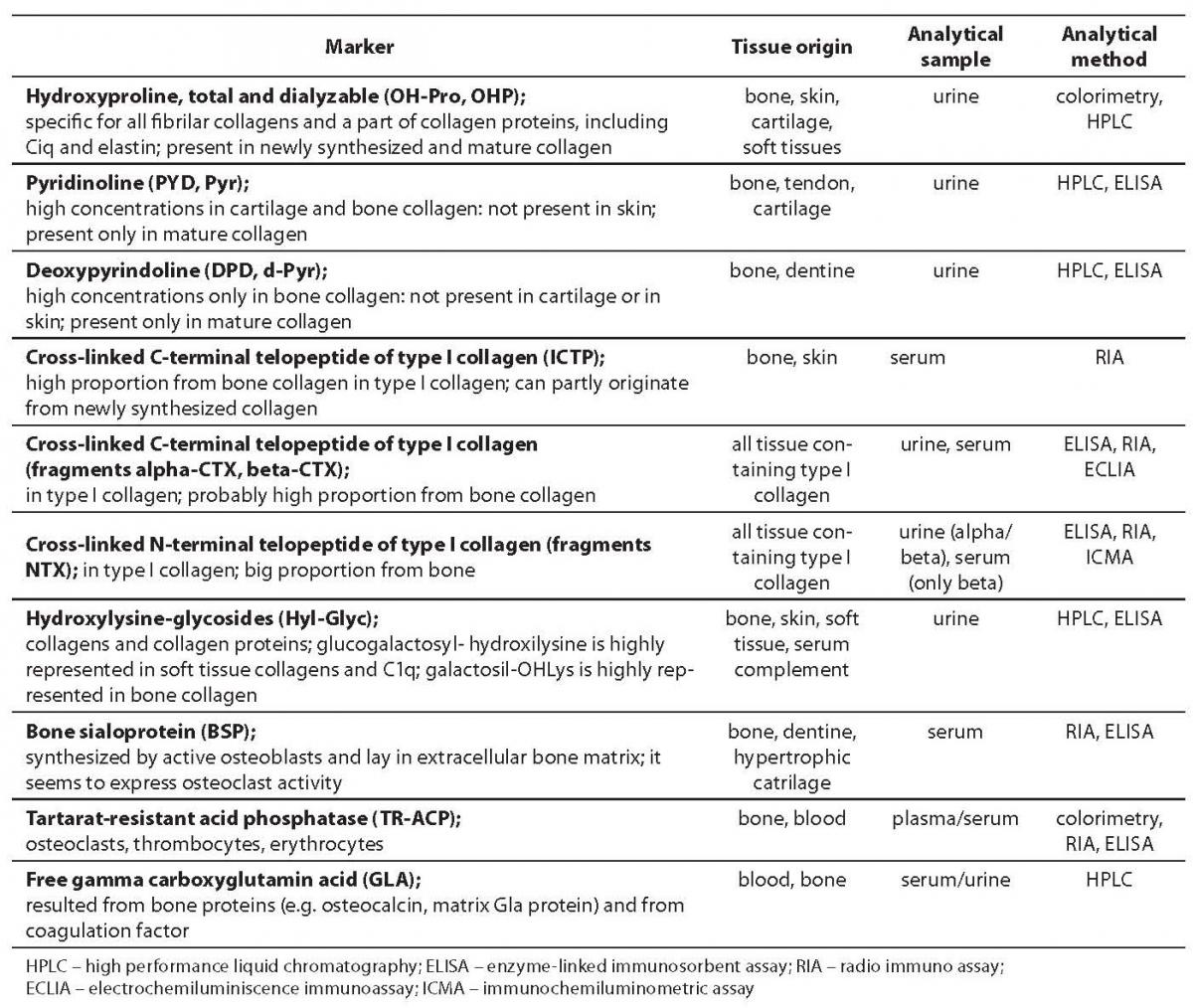
Tables 1 and 2 show bone formation and resorption markers discovered so far for groups a) and b), some of which will be further discussed. Tables 1 and 2 also list the tissue origin of markers, their physiological sample which is measured for their concentration/activity, and currently available analytical methods (1,2). In this review we shall discuss the markers whose standardization is currently underway and which, according to past test results, have shown considerable clinical validity. We will also point out disadvantages of some of hitherto frequently used markers.
Table 1. Bone formation markers
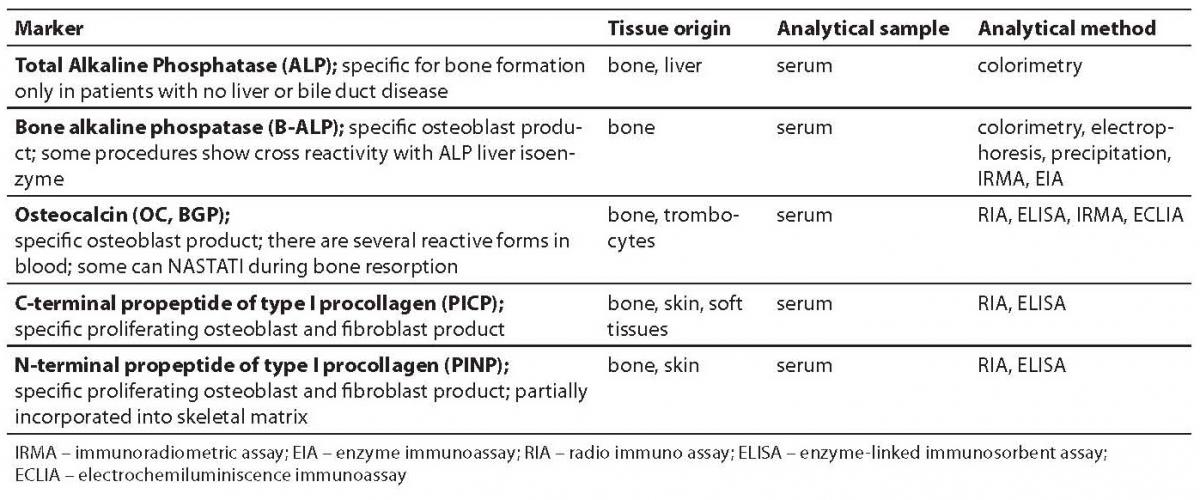
Table 2. Bone resorption markers
What is the general significance of biochemical markers of bone remodeling?
The familiar basic advantage of biochemical markers, compared with bone mineral densitometry and static histomorphometric analysis of bone biopsy, is that by using them we get the information about the status of bone remodeling. In addition, biochemical markers are non-invasive with regard to dynamic histomorphometric analysis of bone biopsy.
Information on bone remodeling status could be an early indicator of some pathological changes in bones or the risk of some bone disease. By measuring concentration/activity of biochemical markers, it is possible to gain information about therapeutic response faster than by measuring bone mass. Significant changes in biochemical markers can be discovered already after 1 to 3 months of effective therapy while bone mass changes can be adequately evaluated only after the 1st or rather the 2nd year (3,4). The need for specific biochemical markers is also emphasized by the use of new, very efficient drugs which have very strong effect on bone metabolism. Together with bone mass measurements, they are also very useful in disease prognosis. It has been shown so far without doubt that some of the markers or marker combinations presented in tables are useful in population and epidemiological studies, as well as in monitoring of anti-resorption therapy effect. However, given the continuous development of new, specific procedures for their measurement, the final assessment of their clinical usefulness in patient management is still underway. Therefore, it is still recommended to pay maximum attention to the issue of whether and how the markers are to be used and to standardization of preanalytical and analytical procedures of bone marker determination.
How to evaluate the clinical validity of markers?
Generally, biochemists and clinical specialists should ask themselves: which biochemical marker of bone remodeling to measure,how and when to measure it, how to collect the sample and how to interpret the test result. Important criteria in critical judgement on whether to measure some bone formation or bone resorption markers are the following:
Biological factors (like tissue specificity, effect of change in liver or kidney function on marker clearance, biological rhythm of the marker due to standardization of physiological sampling time, immobilization, etc.):
Pre-analytical factors (sample storage procedures, i.e. time and temperature, sample freezing and thawing, anticoagulant effect, etc.);
Analytical specificity and accuracy (microheterogeneity of markers as, e.g., degree of ALP glycosylation, possibility of marker resorption into several different fragments as in case of OC, bias in methods due to non-harmonized calibrations, specificity of antibodies and of, e.g., enzyme activity inhibitors);
Diagnostic validity (or the question of differences between markers considering their diagnostic sensitivity and specificity) (1,5).
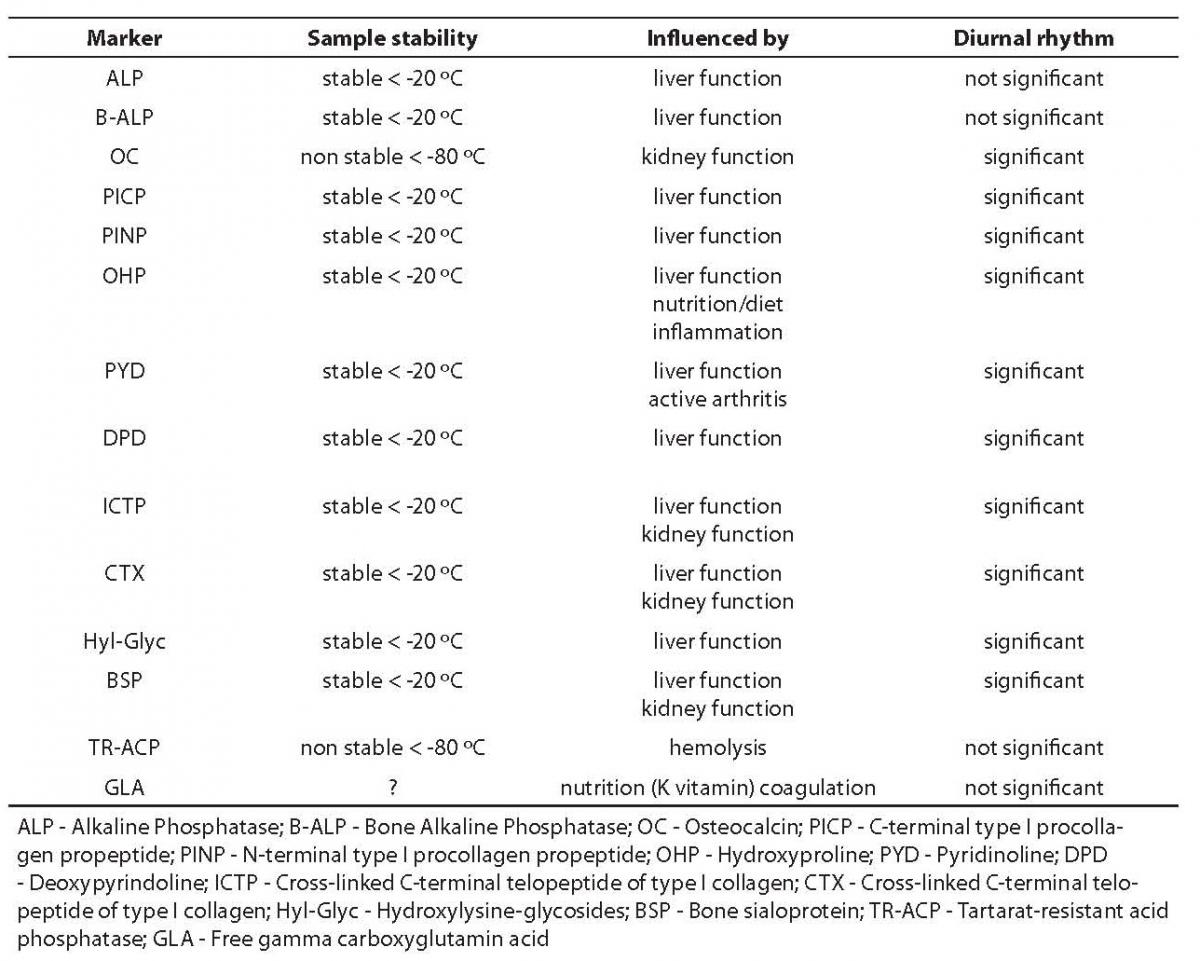
In this context, the review of bone markers to follow should enable the reader to assess the possible diagnostic value of certain markers that have already been subjected to partial evaluation in scientific and professional literature ŠALP (alkaline phosphatase), B-ALP (bone alkaline phosphatase), OC (osteocalcin), PICP (carboxy-terminal type I procollagen propeptide), PYD (pyridinoline), DPD (deoxypyridinoline), CTX (C-telopeptide of type I collagen) and NTX (N-telopeptide of type I collagen)Ć, and to evaluate disadvantages and limitations of procedures applied to measure them. Some of the factors for judgement of bone markers listed above are shown in Table 3 (1,2).
Table 3. Preanalytical and biological characteristics of bone markers
Bone formation markers
Bone formation markers are direct or indirect products or enzymes of active osteoblasts, their concentration or activity is measured in serum or plasma, and generally they are characterized by moderately expressed biological variability. They most often include the measurement of total ALP activity, B-ALP activity or mass, and OC and procollagen type I propeptide concentrations.
Alkaline phosphatase (ALP)
Precise physiological function of ALP is still unclear, but it is assumed to play a role in osteoid formation and bone mineralization. Physiological forms are coded by 4 genetic loci, three tissue-specific and one non-tissue-specific. Bone and liver forms of ALP, the most frequent in serum, are formed by post-translation modifications, or more precisely by different glycosylation stages of tissue-non-specific gene product (different ratio of sialic acid and attached sugar residues). Like ectoenzyme tetramer, ALP is specifically, through C-terminal glycan phosphatidyl anchor domain, attached to osteoblast membranes. In circulation, there is normally only one dimer ALP present, but other tetramer forms can be also found attached to the membrane. By in vitro examinations, some other forms of B-ALP were found that are the result of protease and phospholipase C and D activity (1,2,6,7).
In the serum of healthy adults, approximately 50% of total ALP activity originates from liver, and around 50% from bones, whereas B-ALP prevails in serum from children and adolescents. Total ALP values are higher in men than in women presumably due to a down-regulating effect of estrogen on bone metabolism, and are linear increasing with age in both genders. In women using contraceptives, B-ALP shows 13% less activity. Zinc and magnesium deficiency, which may occur due to parenteral nutrition without microelements, decreases ALP activity in serum. Total ALP activity in serum remains stable for 7 days at room temperature. Repeated fivefold sample freezing and thawing do not influence B-ALP activity (7).
In activity/concentration differentiation and measurement procedures of two most frequently represented isoenzyme ALP forms (liver and bone), we use differences in physico-chemical characteristics of a certain isoform (thermal denaturation, electrophoresis, precipitation, selective inhibition, HPLC and immunochemical procedures).
By immunochemical procedures, it is possible to measure B-ALP mass concentrations using two monoclonal antibodies against B-ALP and activity after immunoadsorption on microtiter plates. Both procedures show cross reactivity of 14-20% with liver form of ALP. However, it has been found that B-ALP value remains within the reference interval in patients suffering from hepatobiliary diseases if total ALP activity in serum does not exceed more than twice the upper limit of the reference interval.
In precipitation procedure, carbohydrate (oligosaccharide) part of B-ALP, rich in N-acetylglucosamine and N-acetylneuraminic acid, precipitates with lectins from wheat seeds, whereas liver ALP does not precipitate and is measured in the supernatant. This method gives false increased test results in patients suffering from hepatobiliary diseases. Normally, biliary ALP can be precipitated with lectin (which is prevented by adding Triton X100 to the reagent) and, due to increased liver cell permeability, we can measure the glycosylated part of liver ALP. Considering the fact that lectin preparation from one bottle to another can vary significantly, B-ALP is in fact not always completely precipitated; therefore, this procedure should be standardized according to the standard reference method. B-ALP activity mass ratio (estimated by precipitation with lectin and IRMA), as well as activation energy (reaction catalyzed by B-ALP) vary depending on examined patient group and include probably different glycosylation stage of enzyme molecules.
Sequential heat-induced denaturation procedure of bone isoenzyme (56 °C) is nowadays almost abandoned.
Although electrophoretic methods for separation of ALP isoforms on different carriers are difficult and require experience, they are still in use due to other advantages. Actually, it is possible by usage of electrophoretic methods to discover also other ALP isoforms and main bone ALP form together with its variants (with concurrent detection of the anchor-carrying form). When B-ALP activity exceeds 50% of the total enzyme activity in serum, neuraminidase-treated sample is required (it removes sialinic acid from B-ALP) or sample application to other lectin containing gel (it precipitates B-ALP). In metabolic bone diseases, three B-ALP peaks were measured using HPLC (1,2,6,7).
In cases when liver disease is excluded, total ALP activity has a clinical significance and provides good information about bone formation and a number of active osteoblasts. Numerous examinations (bone marrow transplantation, prostate cancer with metastases in bones, women in postmenopause, antiresorption therapy monitoring, etc.) have shown that B-ALP measurements have more important discriminating value than the value of total ALP activity, pointing out the higher diagnostic specificity of B-ALP measurements. Therefore, it can be concluded that B-ALP measurement makes clearer difference between “normal” and “pathological” states with the upper reference range values, and thus increases diagnostic specificity in bone diseases. Due to cross-reactivity in immunochemical methods and their inaccuracy in the lower half of the reference interval, electrophoresis remains the gold standard for detecting decreased B-ALP concentration/activity and for confirmation of increased B-ALP in cases of severe liver diseases (6,8,9,10).
Osteocalcin (OC)
Osteocalcin or bone Gla-protein (glutamic acid) is the main non-collagen protein of bone matrix, which is primarily synthesized by osteoblasts and odontoblasts and hypertrophic chodrocytes. Slight quantity of OC may be released during bone resorption and measured by some methods, so it could also be called a bone remodeling marker. It contains 49 aminoacids (5.8 kDa) of which there are three gamma-carboxyl glutamic acids (post-translational, K vitamin dependent enzyme carboxylation) at positions 17, 21 and 24 and they are responsible for calcium-binding characteristics of this protein. The exact role of this protein still remains unclear. Its role in bone mineralization process is discussed, as well as its messenger role for calcitriol in bone resorption and its role as an inhibitor of leukocyte esterase and growth factor activity. After synthesis which is significantly stimulated by calcitriol, OC is secreted and incorporated in skeletal matrix (>80%). One part (10-30%) of newly synthesized OC is released into circulation where its concentration can be immunochemically measured (it reflects OC synthesis in osteoblasts and bone formation). The important heterogeneity of circulating OC fraction is determined due to the fact that it is proteolytically cleaved in liver, kidneys, plasma and bones. There are two main sites of enzymatic cleavage of “intact” molecules, and the resulting known fragments in circulation are: 1-19, 20-43, 44-49, 1-43 and 20-49. In seemingly healthy individuals, the circulating fraction of “intact” OC represents only 30% of total immunoreactive OC, N-terminal/middle regional fraction (1-43) 30%, and other fractions are present in very low concentrations (1,2). Clinical studies showed that vitamin K deficiency may lead to impairment in the carboxylation of OC, also resulting in a disproportionate increase in the undercarboxylated OC form in the circulation (11).
Because it is very quickly secreted through kidneys, the half-life of circulating OC is around 4-5 minutes (OC and its fragments accumulate and their concentration in serum increases when kidney function is changed). Circulating OC shows circadian rhythm with the highest values in the night and early in the morning and the lowest during early morning hours, with the differences reaching up to 50%. In women who take oral contraceptives, the circulating OC values are 24% lower. On the other hand, it has been established that OC values are not influenced by menstrual cycle or calcium consumption through nutrition. It is interesting that OC is the only bone formation marker whose values increase significantly after long-term bed rest, probably due to release of OC incorporated in bones from resorption sites. Also, only the values of OC, and not B-ALP or PICP, are increased in individuals with increased physical activity. In comparison to B-APL, significant analytical and biological OC variants have been described. OC values are higher in children than in adults, especially during the intensive growth period. They are also higher in men, and in women these values are considerably higher during menopause (2,6,12). The concentration of undercarboxylated OC correlates with bone mineral density at the femoral neck (13) and it is an independent predictor of hip fracture in a population of ambulatory elderly women (14).
Concentration of immunoreactive OC in human samples is measured by immunochemical methods (RIA, ELISA, IRMA, ECLIA). Current available methods for the determination of circulating OC differ according to measurement principle (competitive, immunometric), source (polyclonal, monoclonal), antibody specificity (e.g. for “intact” molecule or different fragments) and calibrator origin (bovine/human). Different antibody specificity for OC fragments in these methods results in great differences in immunoreactive OC concentrations (this is especially important in diseases characterized by the accumulation of different fragments as, e.g., chronic kidney disease and Paget’s disease). Poor correlation is observed in method comparability studies. In most cases, these methods measure 1-43 fragment (N-terminal/MID fragment; partially it can be formed by an active osteoblast), a product of proteolytic resorption of intact OC. Although we know little about the function of this fragment, its partial measurement eliminates the problem of preanalytical instability. Actually, loss of immunoreactivity is recorded already after the sample is at room temperature for an hour. Therefore, fast sample processing after sampling is considered to be essential for all currently available methods.
The sample for OC concentration measurement is normally serum, although for some methods plasma can also be used. Due to confirmed instability and protease action, it is recommended to put the sample immediately on ice, e.g. inhibitor aprotinine can be added, thus preserving stability for five hours at room temperature. Serum, separated from cells within the first hour after sampling, can be immediately frozen. However, repeated freezing and thawing are not recommended. Hemolysis can affect OC concentration measurement due to increased release of proteases from erythrocytes (2,7,12). Anticoagulants (the case of blood plasma use) with oxalates and fluorides can decrease OC concentration. It is assumed that the reason is not interference in immunochemical method, but increased higher hemolysis caused by these anticoagulants compared with the others. Furthermore, it has been concluded that OC stability depends extremely on analytical specificity of the applied method. Immunometric measurements specifically measure intact OC which tends to accelerate proteolytical resorption in serum. By these methods, lower values (10%) of OC 1-49 and OC 1-43 were found during 7 day storage at +4 °C.
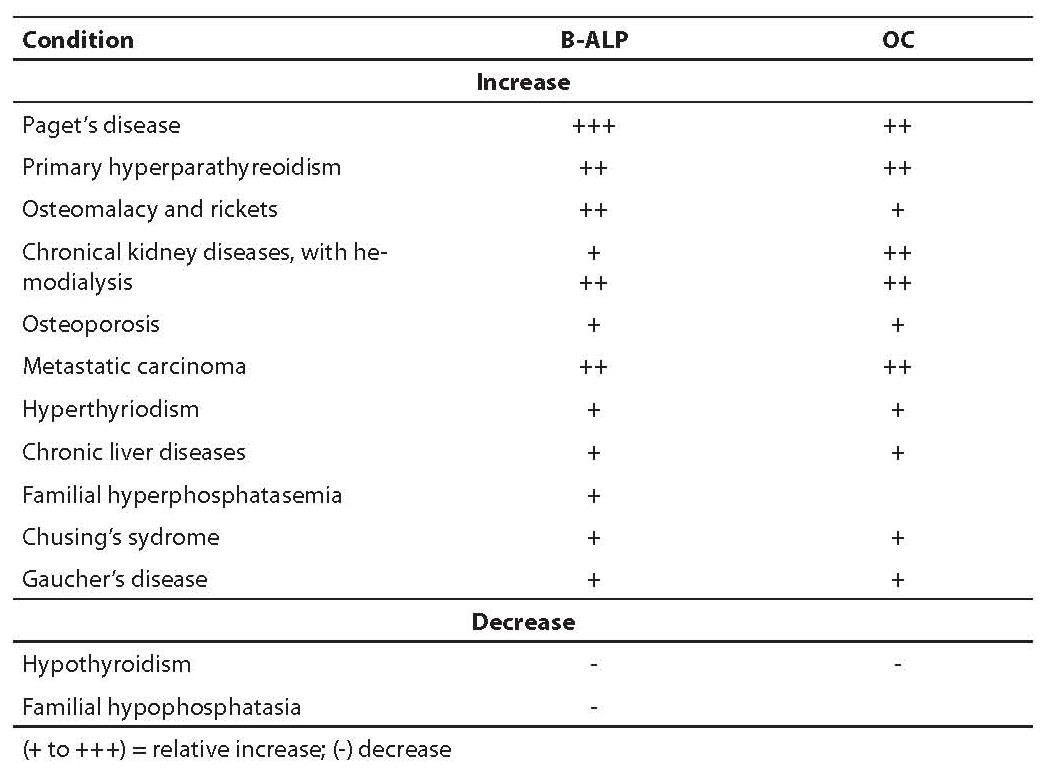
Elevated OC values are described in patients suffering from increased bone formation – hyperparathyroidism, Paget’s disease, significant remodeling due to osteoporosis, hyperthyroidism, renal osteodystrophy, fractures and acromegaly. In women in later post-menopausal period, measurements of OC 1-49 and of OC1-49/OC 1-43 ratio have the same diagnostical significance. It is interesting that in patients suffering from osteoporosis OC and B-ALP correlation is low, while in primary hyperparathyroidism OC and B-ALP concentration show comparable discriminating value (9). In patients having bone tumor and metastases and those with Paget’s disease, OC measurements are less important than B-ALP measurements regarding diagnostic sensitivity. On the contrary, OC determination is more important than B-ALP measurement in monitoring patients under corticosteroid therapy. In patients with chronic kidney diseases, the usefulness of OC is significantly decreased because it is influenced by renal function. Decreased OC values were found in hyperparathyroidism, hypothyroidism, growth hormone deficiency, during estrogen substitution therapy and therapy with glucocorticoid, biphosphonate and calcitonin. Table 4 shows clinical conditions connected to changes in B-ALP and OC in serum. For appropriate data interpretation of OC concentration, it is essential whether the applied measurement method is based on reaction of OC fragments released from bone matrix during resorption. Only in case of no reaction with these fragments is it justified to measure bone formation markers repeatedly (OC 1-43 is a fragment that is probably not released from the bone during resorption, but only from a newly synthesized OC). The measurement of specific OC fragment is more promising since intact molecule determinations are subject to preanalytic instability.
Table 4. Clinical conditions connected to changes in B-ALP and OC values in serum
Type I propeptid procollagen (PINP, PICP)
Type I collagen is the main protein of bone matrix (>90% matrix content) and to a lesser extent of skin, dentine, tendon, cornea and other tissues. It is synthesized in osteoblasts as a precursor of procollagen I that contains N- and C-terminal trimeric extended domain Štwo identical polypeptide chains (alpha1 I) and one (alpha2 I)] known as propeptide (PINP and PICP). Characteristics of the two mentioned propeptides are shown in Table 5 (1).
Table 5. Comparison of propeptide type I procollagen
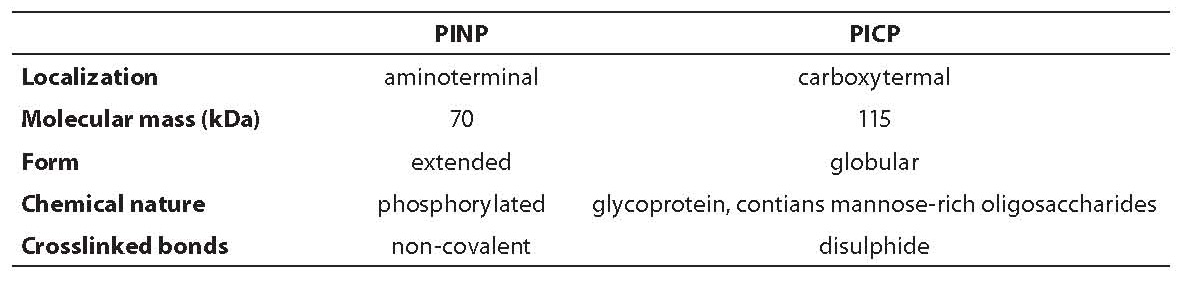
Procollagen is, therefore, 50% longer molecule than the final protein, its function is to prevent a precocious aggregation of collagen molecules in fibrils inside the cell. Before collagen fibril maturation, these so-called C- and N-propeptides are cleaved from procollagen type I by specific extracellular tissue endopeptidases. C-terminal propeptide type I procollagen (PICP) is a glycoprotein that contains two polypeptid chains (alpha1 I) of 246 and one polypeptide chain (alpha2 I) of 247 amino acid residues, with intra- and intercellular disulphide bonds (115 kDa) (1,6,12,15). Carbohydrate component of C-terminal propeptide contains N-acetylglucosamine and mannose residues. PICP is metabolized through mannose-6-P receptor on liver endothelial cells (half-life of 6-8 minutes) and PINP through scavenger receptor. Every liver dysfunction might result in variation of PICP liver clearance, or elevated concentration in circulation. In contrast, proinflammatory cytokine action, which regulates endocytosis through sinusoidal cells, can result in low PICP values. Significant genetic influence on PICP values is also described, as well as diurnal rhythm with amplitude of 20% (higher values during the night and lower in the afternoon) and relative sample stability (15 days at +4°C, several months at -20°C). PICP is therefore not incorporated in bone matrix but is released into circulation, where it can be measured by different immunochemical methods (RIA, ELISA). It is considered that the process of PICP formation in other tissues lasts longer; therefore, it is thought that it contributes a little to a circulating pool, although this information is not completely clear.
In primary hyperparathyroidism, PICP values are within reference value limits in healthy individuals (while, e.g., ALP and OC values are elevated). In patients suffering from osteomalacy on D vitamin supplementation, elevated values were recorded also after paraidectomy. Lowered PICP values were found in patients with osteogenesis imperfecta and in individuals receiving glucocorticoid therapy (1,2). Generally, through examinations on different clinical models, it appears that PICP is less sensitive and specific than B-ALP and OC due to relative bone non-specificity and different metabolic clearance.
Bone resorption markers
Aside from TR-ACP (tartrate-resistant acid phosphatase), bone resorption markers are resorption products of bone collagen. As they are secreted in urine, they were mostly measured in urine until recently; however, samples had significant test result variability. Therefore, the main scientific and commercial interest has been focused on setting up and evaluating methods for their measuring in serum.
Collagen cross-links (PYD, DPD)
With a series of intra- and intermolecular covalent bonds (cross-links) between terminal nonhelical portion of the molecule and helical portion of a neighboring collagen molecule, a molecule of mature collagen type I is stabilized in skeletal matrix. Cross-links, as a 3-hydroxypyridinium ring, are formed by deamination of lysine or hydroxylysine epsilon-amino group with catalytic action of enzyme lysyloxydase. There are two nonreducible cross-links identified in human urine: deoxypyridinoline (DPD) which is formed by reaction of side-chains of two hydroxylysine molecules and one lysine molecule, and pyridinoline (PYD) which is formed by reaction of side-chains of three hydroxylysine molecules (both compounds have inborn immunogenetics and fluorescence) (2,6,7,12,16). DPD is found mostly in bones, not so much in dentine, while PYD is located in bone collagen fibrils and cartilage and to a lesser extent in other tissues (tendons, ligaments, blood vessel walls). Since the bones have the most intensive remodeling, they are considered as the most important PDP and PYD source. When collagen matrix is proteolytically degraded, both cross-links are released into circulation and, due to their small mass, they are secreted by urine where they can be measured. Both types of cross-links are secreted as free (40%) and peptide-linked (60%) amino acid derivatives. Some biological factors that can influence DPD and PYD values have been described: circadian rhythm indicates maximum values between 5 and 8 a.m. and minimum between 5 and 8 p.m.; menopause, osteopenia or long-term bed rest do not affect the values; after a 15-month monitoring period, intraindividual variability between 20 and 30% has been recorded: in men the linear ratio between age and secretion of these compounds through urine is high, while such clear dependence has not been recorded in women; during pregnancy the values are elevated for about 91% comparing the first and the last trimester. DDP is considered a specific resorption marker because it is formed during collagen maturation (not during biosynthesis and therefore it appears only as a resorption product of the mature matrix), and it does not metabolize before secretion into urine. The main source of DPD is bone, and it is not absorbed from food (1,2,16,17).
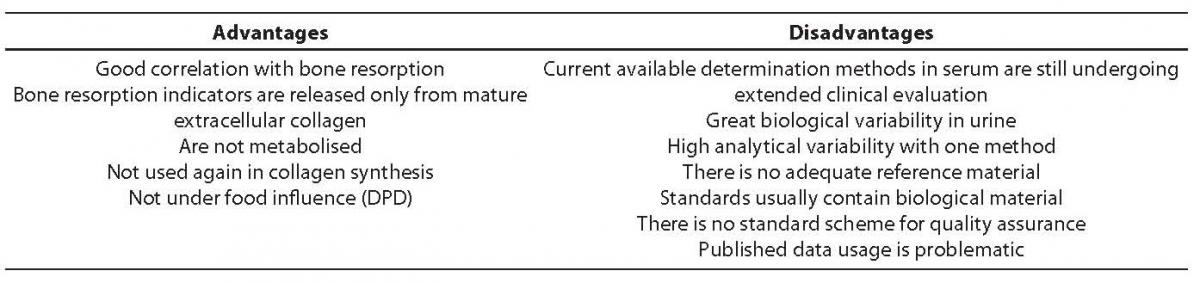
DPD and DYP concentration measurement can be performed using HPLC, fluorometric quantification and immunochemical methods. With ELISA, we can specifically measure free, non-linked and peptide-linked DPD form. Thus attained DPD and DYP values show high correlation (R > 0.95) with the values measured using HPLC, a method considered agold standard. The second morning urine (between 8 and 10 a.m.) after a 12-hour fast is the recommended sample. Exposing the sample to the UV light causes fast resorption (half-life < 30 seconds) of both cross-links, while normal daily light does not show so strong effect. Marker stability at -20°C is 10-20 years. No significant value decline is recorded if samples are stored at temperature <20°C during a period of 6 weeks. Repeated sample freezing and thawing do not affect DPD and DYP values. Table 6 shows advantages and disadvantages of collagen cross-links as bone resorption markers.
Patients with non-treated primary hyperparathyroidism show significantly elevated cross-link values which correlate with total ALP activity and PTH concentration. Tumor patients with and without metastases in bones can be differentiated by simultaneous B-ALP and immunoreactive PYD measurements with 0.89 accuracy (ROC analysis). Patients with D vitamin deficiency show triple increase in cross-links; PYD and DPD value comparison between premenopausal healthy women and postmenopausal (osteopenic) women shows increase in urinal cross-link secretion by up to 105%; urinary PYD is increased in 40% of patients suffering from Paget’s disease, while, e.g., total ALP activity lies within the reference interval; elevated PYD values in serum have been determined also in patients with renal osteodystrophy and correlate with histomorphometric indicators of bone resorption.
Table 6. Advantages and disadvantages of collagen cross-links
Hidroxyproline (OHP)
Considering characteristics of newer, more specific bone resorption markers, it is not recommended to measure traditionally used biochemical hidroxyproline (OHP) marker because of the following disadvantages: OHP is also recorded in collagens of other tissues, in C1q, elastin and acetylcholinesterases; it is not a specific resorption marker because it is also released during extracellular metabolism of newly synthesized (pro)collagen; 90% of OHP is metabolized by oxidation in liver; OHP secretion in urine is extremely dependent on collagen content in nutrition.
Telopeptides of type I collagens (CTX, NTX)
Theoretical basis for measuring telopeptide collagen regions, instead of collagen cross-links, is the fact that cross-links always involve specific molecular domain, the so-called C- or N-terminal telopeptide. During osteoclast-mediated resorption of type I collagen N- and C-terminal telopeptide fragments, still attached with cross-links to the helical fragment of neighboring molecule, these are released into circulation and eliminated by kidney filtration (1,6,7,18,19). Thereby is created an excess of telopeptide resorption products due to the following reasons: cleavage of polypeptide chains can occur at several sites in a telopeptide which can or does not have to be cross-linked, there can be several types of cross-linking, one or more telopeptide chains can be altered by beta-isomerization, and telopeptides can be cross-linked to helical collagen region. For some of these resorption products, measurement methods are available. There is an immunochemical method for measurement of C-terminal telopeptide in urine or, more precisely C-telopeptide resorption product that uses monoclonal antibodies against synthetic octapeptide that has a cross-linked site (Glu-Lys-Ala-His-beta-Asp-Gly-Gly-Arg) called beta-CTX or beta CrossLaps).
Urinary concentrations are elevated in more than 1/3 of early postmenopausal women. After hormone replacement therapy, values decrease significantly (up to 61%). It is possible to measure nonisomerized urine octapeptide (alpha-CTX) and simultaneously to measure alpha/beta CTX as a bone remodeling index (the index is increased in Paget’s disease, and decreased after biphosphonate therapy). Another method is the immunochemical method for CTX measurement in serum that uses monoclonal antibodies specific for isomerized sequence (EKAHD-beta-GGR) from alpha-1 chain of human type I collagen (2,6,7). However, lipemic serum can interfere with this method, the samples stored at room temperature or at +4 °C have lowered values by up to 13%, and repeated freezing gives results with lower concentration (10%). According to present studies, CTX values show high specificity (100%) and sensitivity (83.8%) in monitoring the response of anti-resorption therapy (after 6 months over 92% women on anti-resorption therapy). CTX concentrations adequately express bad prognosis in multiple myeloma, they are connected with more severe radiography test results in patients suffering from rheumatoid arthritis. High CTX values are related to low bone mass in patients suffering from Crohn’s disease and show high efficacy in diagnosis of bone metastases. Methods for telopeptide N-fragments (NTX) are commercially available. The antibodies recognize conformational epitope of cross-linked alpha-2-N-telopeptide with a certain sequence (QYDGKGVG) which is a product of osteoclastic proteolysis. Circadian rhythm is described, and it is interesting to find that there is an increased concentration during the follicular period which declines during the luteal phase in women. Also, concentrations increase with age and are higher than those in the respective male age group. Present results show that this marker could be significant in bone resorption assessment because N-terminal telopeptide in the urine of children displays growth rate, its concentration is significantly elevated in (female) patients with postmenopausal osteoporosis, indicating an adequate suppression of bone remodeling through estrogen supplementary therapy. In individuals taking biphosphates, the urinary excretion of N-terminal telopeptide is a better indicator of bone remodeling than collagen cross-links.
Tartarate-resistant acid phosphatase (TR-ACP)
Acid phosphatases are lysosomal enzymes of different tissue origin (platelets, erythrocytes, bone, prostate) that hydrolyzes phospho-monoesters at low pH value. Five isoenzymatic forms of enzymes have been found in plasma that differ regarding tissue and chromosomal origin, as well as molecular mass and electrophoretic mobility. By electrophoretic mobility, they are classified as isoenzymes 1-5, and according to sensitivity or inhibition with L(+)- tartarate; they are furthermore classified by tartarate-sensitive and tartarate-resistant forms (6,11,20,21,22). Total TR-ACP activity measurement in serum, classified as a bone resorption marker, has many disadvantages (relatively low activity, presence of inhibitors, instability by alkaline pH, interference by hemolysis) and it is, therefore, not recommended for diagnosis.
Type 5 TR-ACP is formed by macrophages (TR-ACP 5a; activity enhanced in Gaucher’s disease and hairy cell leukemia) and osteoclasts (TR-ACP 5b; non sialyl residues; increased activity in bone diseases, especially in osteoporosis). Type 5b TR-ACP changes reflect a number of active osteoclasts.
The majority of TR-ACP activity measurement methods cannot distinguish between osteoclast enzyme form (isoform 5b) and other forms in plasma. Two recently set up measurement methods are being evaluated. These are kinetic measurement procedure where TR-ACP inhibition with fluoride and TR-ACP 5a with heparin is used, and immunechemical method that uses monoclonal antibody for TR-ACP 5b.
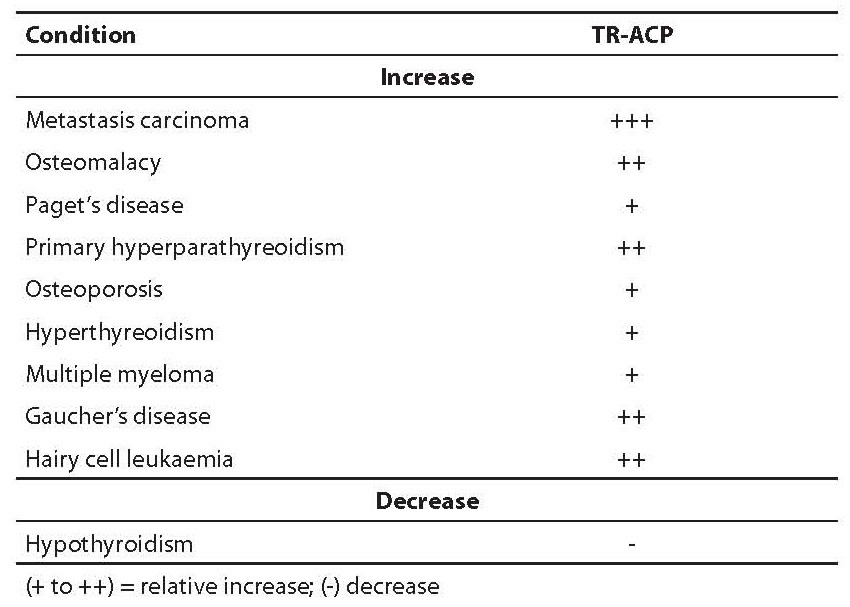
TR-ACP activity is increased in patients with different diseases, as shown in Table 7, especially in bone metastases. It is also considered as a second class bone resorption marker, especially in monitoring patients suffering from Paget’s disease who received biphosphonate therapy.
Table 7. Clinical conditions associated with changes in TR-ACP activity
Total activity measurement of TR-ACP as a bone resorption marker is currently not recommendable given the low enzyme activity and inhibitor presence in serum, instability at alkaline pH, the fact that L(+) tartarate is a competitive inhibitor, and strong interferential hemolysis.
Hydroxylysine-glycosides (Hyl-Glyc)
Galactosyl hydroxylysine (GHL) and glucosyl-galactosyl-hydroxylysine (GGHL) both originate from lysine during posttranslatory phase of collagen synthesis and are released into circulation during collagen resorption. GHL is relatively more specific for bone matrix resorption than GGHL, and without doubt more specific than, e.g., OHP. The real advantage of Hyl-Glyc as a resorption marker is the fact that these forms are not metabolized and are not affected by nutrition factors. In urine, they can be measured by HPLC. It is recommended to set up an appropriate immunochemical method and to make final evaluation of this marker for clinical significance (1,6,12).
Other bone matrix proteins
There is a small number of published scientific data about two matrix protein that seem to have certain potential as bone resorption markers.
Osteonectine is secreted as Ca-binding glycoprotein found in different cells, including osteoblasts, endothelial cells and fibroblasts. It is present in active osteoblasts and young osteocytes (but not in inactive osteocytes) and therefore it is considered suitable as a marker for differentiation of osteogenetic bone cells indicating bone formation. However, due to its presence in the majority of connective tissues and in platelets, its role as a circulating marker is diminished.
Bone Sialoprotein (BSP) is a phosphorylated glycoprotein, significantly posttranslationally transformed and synthesized by osteoblasts and ondoblasts. In contrast to other bone matrix phosphorylated glycoproteins (osteonectin), BSP is relatively limited to the bone. It stimulates in vitro formation of hydroxyapatite and is assumed to act as a cell adhesion molecule, enabling cells (osteoclasts) to attach to extracellular matrix. Currently there are only available preliminary test results on evaluation of this bone remodeling marker (ELISA) in patients suffering from the early stage of rheumatoid arthritis (elevated values in serum), as well as in synovial fluid of patients suffering from rheumatoid arthritis with progressed joint function disorder (higher values were found than in patients with preserved joint structure). Elevated concentration of circulating fractions in postmenopausal women is also noticed in comparison to reference values in premenopausal period (1). As a good indicator of possible fracture and bone mass, cathepsin K has also been mentioned, which is a cysteine protease important in the bone resorption process (23,24,25).
Based on the published scientific data review, the following can be concluded:
B-ALP activity measurement has an advantage compared to the measurement of total ALP activity because of greater diagnostic sensitivity and specificity. Immunochemical methods for B-ALP measurement show cross reactivity (14-20%) with liver ALP. However, this does not diminish clinical efficacy of these measurements, except in patients with severe liver diseases.
OC is a bone specific protein but it shows numerous disadvantages like instability in the sample and differences in test results due to different measurement methods. However, in some diagnostic situations (corticosteroid osteopenia, lack of destroyed bone structure) OC can be used as a sensitive bone remodeling marker.
The urinary excretion of collagen cross-links is partly evaluated analytically but, based on its clinical efficacy; it is considered that this marker can replace OHP measurement as the method of choice for bone remodeling evaluation.
Other telopeptide type I collagen resorption products (NTX, CTX) are measured as bone resorption indices; however, they demand further evaluation, especially of their extraosseous clearance and other possible extraosseous sources. Their clinical efficacy varies depending on which patient group is examined.
TR-ACP activity measurement is not recommended for clinical use.
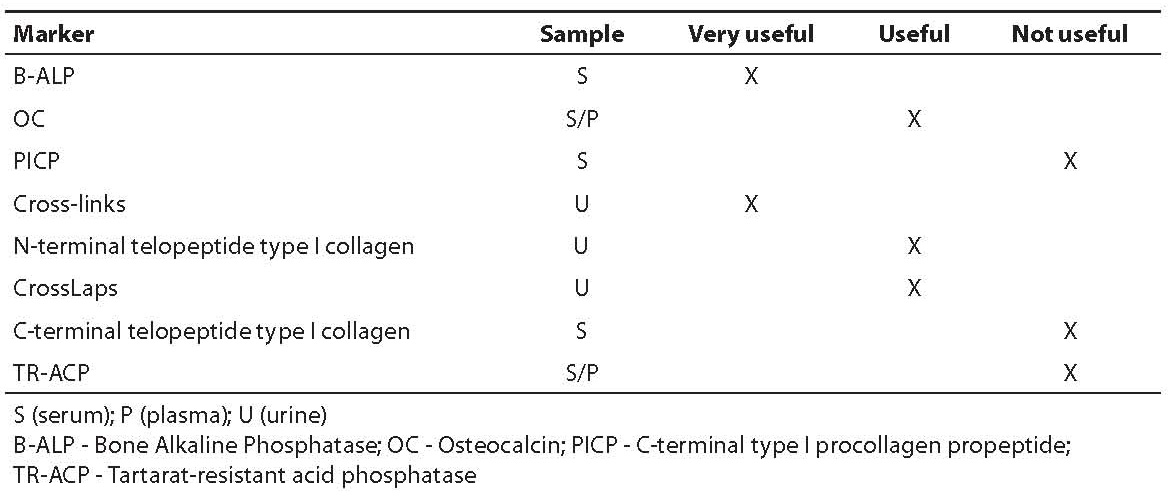
Table 8 summarizes the evaluated clinical efficacy of bone remodeling markers.
In order to select a bone remodeling marker, it is important to do inter-laboratory comparisons and to define the characteristics of the bone marker to be used; to establish reference intervals, to standardize measurement methods, to establish a good quality assurance program, to try to diminish individual variability by measuring markers in serum and not in urine, to use automated measurement methods with appropriate calibration, and to test the stability of reagents.
Considering the frequency of osteoporosis cases in a wider population and different types of antiresorption therapy, International Osteoporosis Foundation (IOF) published in 2000 some guidelines to use on various types (bone formation markers B-ALP, OC and PINP in serum; bone resorption markers NTX in urine, CTX in serum and urine, DPD in urine) and number of bone markers, sampling and measurement intervals of bone marker concentrations during therapy periods, and limiting values considering risk assessment for fracture and possible malignancy stages. These guidelines recommend the use of one bone marker or one bone formation and one bone resorption marker, with the priority given to the measurement of a marker in serum. Blood should be collected after an overnight fast, before 9 a.m.; if a marker is measured in urine, it is recommended to use 1st or 2nd morning urine sample. It is recommended to measure bone formation markers before and 6 months after therapy, and bone resorption markers before and 3 or 6 months after therapy (25).
Table 8. Critical evaluation of clinical efficacy of bone remodeling markers
Conclusion
To summarize, there is a need for long-lasting longitudinal studies of biochemical bone remodeling markers on a great number of individuals with correlation to reference procedures of bone mass measurement. Furthermore, rationalization in marker usage or their combination will be essential in order to eliminate those with low clinical value. Indeed, the greatest challenge of the future is the combination of genetic and biochemical markers in risk assessment of osteoporosis and other bone diseases.