Introduction
The German Pathologist Virchow is credited with suggesting the casual link between inflammation and cancer in the 19th century (1). This conclusion was based on the observation that tumours often developed in the setting of chronic inflammation and that inflammatory cells were present in tumour biopsy specimens. Epidemiological studies have revealed that chronic inflammation predisposes to different types of cancer. It is estimated that underlying infections and inflammatory responses are linked to 15-20% of all death from cancer worldwide (2,3). The triggers of chronic inflammation which increase cancer risk include microbial infections (e.g. Helicobacter pylori for gastric cancer and mucosal lymphoma), autoimmune diseases (e.g. inflammatory bowel disease for colon cancer), and cryptogenic inflammatory conditions of uncertain origin (e.g. prostatitis for prostate cancer). The strongest evidence in humans of the role of inflammation in cancer has been provided by studies showing that long-term therapy with anti-inflammatory drugs resulted in decreased numbers of relapses or fewer appearances of new tumours. Recently, Rothwell et al. (4) re-analyzed patient data from eight randomized trials of daily aspirin taken for prevention of cardiovascular disease. Aspirin users were found to have a significantly lower risk of death from cancer than those who didn’t take the drug (4). Earlier studies reported that daily uses of aspirin and other non-steroidal anti-inflammatory drugs (NSAIDs) over extended periods reduced the risk of colorectal cancer or polyp recurrence, but little evidence was available that aspirin might also reduce risk of other cancers (5). In trials in which the patients took aspirin for at least 7.5 years, the 20-years risk of cancer death, starting from the time the trials began, was reduced by 60% for gastrointestinal cancer and by 30% on average for other solid cancers, such as oesophageal, pancreatic, stomach, lung, brain, and prostate cancers (4). These data indicate that anti-inflammatory drugs prevent both gastrointestinal and other solid-organ cancers, and suggest that an inflammatory component is present in the microenvironment of most neoplastic tissues, including those not etiologically related to an obvious inflammatory process.
Key features of cancer-related inflammation (CRI) include leukocyte infiltration, mostly tumour associated macrophages (TAM); the presence of cytokines such as TNF-α, IL-1, IL-6 or chemokines such as CCL2 and CXCL8; and the occurrence of angiogenesis and tissue remodelling. Molecular and cellular pathways linking inflammation and cancer have been identified (2). Two general pathways can be schematically described. In the intrinsic pathway, genetic events (e.g. oncogenes, genetic aberrations) causing neoplastic transformation, initiate the expression of inflammation-related programmes which guide the construction of an inflammatory microenvironment. The extrinsic pathway is driven by inflammatory leukocytes and soluble mediators that establish inflammatory conditions that increase cancer risk. The chronic inflammation associated with the infections caused by most or all pathogens (e.g. Hepatitis B and C viruses, and Helicobacter pylori) favours initiation and progression of tumours (6). In addition to infections, mechanical, radiation, and chemical insults may be responsible for induction of inflammation associated with human malignancy. Key orchestrators at the intersection of the intrinsic and extrinsic pathway include transcription factors Še.g. Nuclear Factor kappa-B (NFκB), Signal Transducers Activator of Transcription3 (STAT3)Ć (7,8), cytokines (e.g. TNF), and chemokines (9). Thus, cancer-related inflammation is a key component of the tumour microenvironment and a recognized hallmark of cancer (10,11). Here we will review both intrinsic and extrinsic pathways and their mutual connection and influence. Knowledge of these mechanisms may not only enhance our understanding of the process of carcinogenesis and of the multifaceted role played by inflammation, but could also help to identify new targets for therapeutic intervention.
The intrinsic pathway links inflammation and oncogenes
The presence of inflammatory components in tumours not epidemiologically related to inflammation raised the question of whether genetic events that cause neoplasia can be responsible for the generation of an inflammatory environment. This question has been addressed by using preclinical and clinical settings in which various oncogenetic mechanisms can be assessed. A useful clinical example of connection between oncogenes and inflammatory milieu is represented by human papillary thyroid carcinoma (PTC), a tumour characterized by the presence of chemokine-guided macrophage and dendritic cell infiltration (12-14). Rearrangements of the chromosome where is located the gene encoding the protein tyrosine kinase RET represent a frequent, early, causative, and sufficient genetic event in the pathogenesis of PTC. In an appropriate cellular context provided by primary human thyrocytes, the activation of RET induces a transcriptional programme connected to inflammation (12). In particular, the RET/PTC-activated transcriptome profile includes: colony-stimulating factors (CSFS), which promote leukocyte recruitment and survival; interleukin 1β (IL-1β), one of the main inflammatory cytokines; cyclo-oxygenase 2 (COX2), frequently expressed in cancer and involved in the synthesis of prostaglandins; chemokines attracting monocytes and dendritic cells (CCL2, CCL20); angiogenic chemokines (CXCL8); co-ordinate induction and inhibition of matrix-degrading enzymes and inhibitors; up-regulation of L-selectin and expression of the chemokine receptor CXCR4 on the initiated cells (15). Key elements of the RET/PTC-activated inflammatory programme were found in biopsy specimens, and patients with lymph node metastasis showed higher levels of the inflammatory molecules in their primary tumours (12,15,16). These results show that an early, causative, and sufficient genetic event (RET/PTC) involved in the pathogenesis of a human tumour directly promotes the build-up of an inflammatory microenvironment to its direct advantage (12).
More in general, different types of alterations concurring to tumour progression, such as activation of oncogenes, or inactivation of tumour suppressors, may trigger the inflammatory cascade. Members of the epidermal growth factor receptor (EGFR) family have tyrosine kinase activity and are frequently involved in human cancer. EGFR activation in glioma induces COX2 expression via p38-mitogen-activated protein kinase (MAPK) activation of Sp1/Sp3 (17); COX2 is an independent prognostic factor in glioma. Ras family oncogenes, the most frequently mutated dominant oncogenes in human cancer, when activated, are able to induce expression and production of inflammatory mediators. Transfer of ras oncogene into a cervical carcinoma line (HeLa) induces the production of CXCL8 (18,19), a chemokine which promotes angiogenesis and tumour progression. Moreover, mild chronic pancreatitis, possibly mirroring clinical epidemiology, acts in concert with K-rasmutation to induce pancreatic intra-epithelial neoplasia and invasive ductal carcinoma (20). Along the same lines Braf, frequently activated in malignant melanoma, induces cytokines which contribute to a pro-tumour milieu (21). Another oncogene, myc, encodes a transcription factor that is over-expressed in many human tumours: deregulation of this gene initiates and maintains key aspects of the tumour phenotype. In addition to promoting cell autonomous proliferation, myc instructs remodelling of the extracellular microenvironment with inflammatory cells and mediators playing key roles. The myc-activated genetic programme also includes several CC chemokines which recruit mast cells. Mast cells, that have long been known to drive angiogenesis, here sustain new vessel formation and tumour growth (22).
Tumour suppressor proteins can also regulate the production of inflammatory mediators. Examples of such proteins are the von Hippel Lindau/hypoxia-inducible factor (VHL/HIF), transforming growth factor-β (TGF-β) and phosphatase and tensin homologue (PTEN). The chemokine receptor CXCR4, frequently expressed on malignant cells and implicated in cell survival and metastasis, as well as TNF-α lies downstream of the VHL/HIF axis in human renal-cell carcinoma cells (23). In non-small cell lung cancer (NSCLC) mutation of PTEN results in up-regulation of HIF-1 activity and in HIF-1-dependent transcription of the CXCR4 gene, which promotes metastasis formation. In an animal model of breast carcinoma, inactivation of the gene encoding the type II TGF-β receptor induces the production of CXCL5 and CXCL12, attracting myeloid-derived suppressor cells (MDSC) which facilitate metastasis (24,25). Alpha catenin is more than a tumour suppressor sequestering beta-catenin: its ablation results in NFκB activation, induction of genes involved in inflammation, cell proliferation, wound-healing, and ultimately squamous cell carcinoma (26). The putative tumour suppressor semaphorin 3B is also able to trigger an IL-8-mediated pro-metastatic programme, suggesting a multifaced role for this molecule (27). P53 has been connected to CXCR4 ligand expression. Indeed, within both human and mouse fibroblasts, p53 can suppress the production of CXCL12 and attenuate tumour development and metastasis (28).
Thus, oncogenes representative of different molecular classes and modes of action (tyrosine kinases, ras-raf, nuclear oncogenes) as well as tumour suppressor genes, share the capacity to orchestrate pro-inflammatory programmes (Figure 1). Although these responses may share common elements in several tissues (e.g. a link to angiogenesis, recruitment of cells of myelo-monocytic origin) some issues, such as the nature of inflammatory components, if they are essential or redundant, their role in different tissues and in different types of cancer, need to be elucidated.
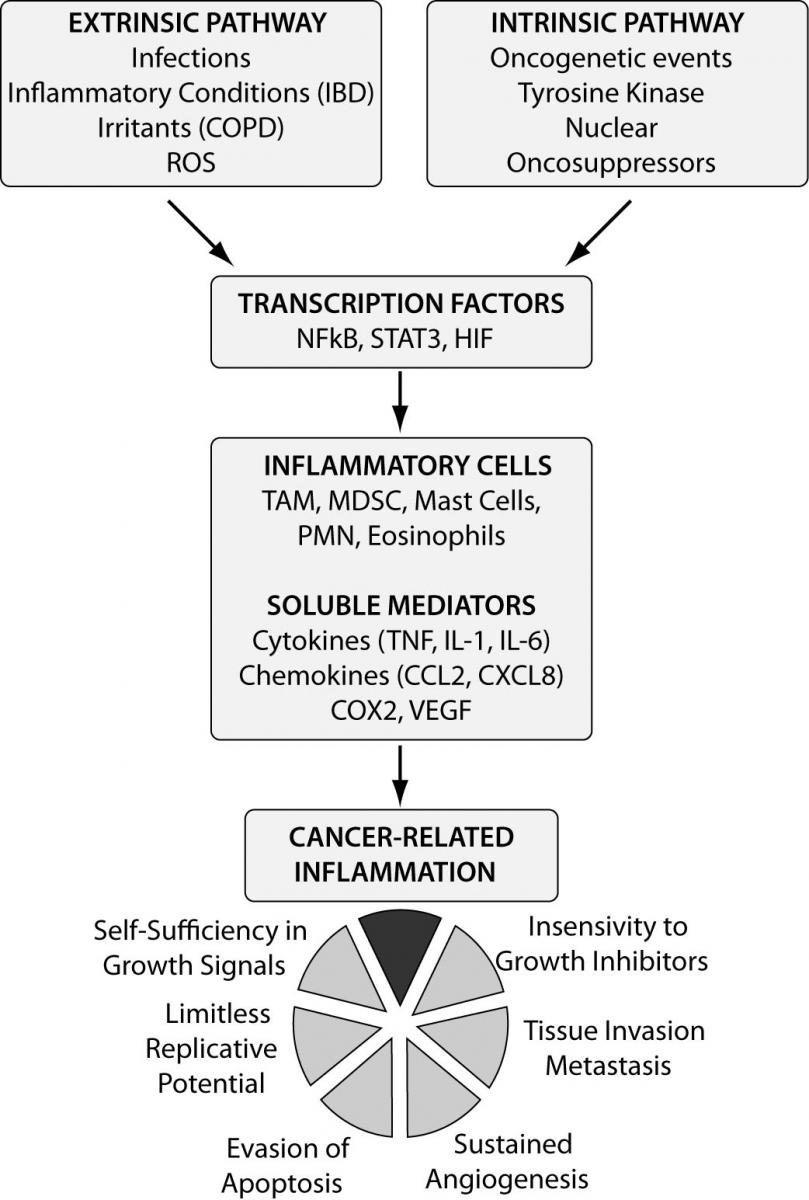
Figure 1. Molecular pathways linking inflammation and cancer. Two pathways can be identified as the major affluent to the inflammatory milieu: the intrinsic one, where genetic events (e.g. oncogenes) inducing neoplastic transformation trigger the inflammatory cascade, and the extrinsic pathway where chronic inflammation (e.g. infections, irritants) significantly increases the risk for different types of cancer. The two pathways converge, resulting in the activation of transcription factors (e.g. NFκB) that coordinate the production of inflammatory mediators and the activation of various leukocytes generating a cancer-related inflammatory microenvironment. CRI represents a recognized hallmark of cancer.
IBD - inflammatory bowel disease; COPD - chronic obstructive pulmonary disease; ROS - reactive oxygen species; NFκB - nuclear factor kappa B; STAT - signal transducers activator of transcription; HIF - hypoxia inducible factor; TAM - tumour associated macrophages; MDSC - myeloid-derived suppressor cells; PMN - polymorphic nuclear cells; COX2 - cyclo-oxygenase 2; VEGF - vascular endothelial growth factor.
The extrinsic pathway
In the extrinsic pathway, inflammation is mainly triggered by leukocytes producing inflammatory mediators. The implicated inflammatory conditions are quite diverse, including a wide array of chronic infections, exposure to noxious agents that trigger inflammation (e.g. gastric acid reflux, tobacco, asbestos) and auto-immune conditions (22,29). The best established link between chronic inflammation and cancer is colorectal cancer that develops in patients with inflammatory bowel diseases (IBD, ulcerative colitis and Crohn disease). These patients have five- to seven-fold increased risk of developing colorectal cancer (43% of patients with ulcerative colitis develop colorectal cancer after 25 to 35 years) (30). In a murine model of IBD, the development of colitis-associated colorectal cancer can be inhibited by blocking TNF-α expression (31). Lung cancer is a leading cause of cancer deaths (32). Although tobacco exposure is evident in nearly 90% of all patients with lung cancer, other chronic airway inflammatory conditions (e.g. asbestosis, silicosis, exposure to airborne particulate matter, idiopathic pulmonary fibrosis, tuberculosis, etc) are all independent risk factors for lung cancer and may account for a proportion of the non-smoking related cases (33).
Reactive oxygen species (ROS) and nitrogen intermediates are obvious inflammation-generated candidate mediators for DNA damage, and evidence obtained in vitro and in vivo is consistent with this view (2). Recently, mitochondrial ROS production has been demonstrated to be required for mediating K-ras-induced lung cancer in mice (34). In addition, mitochondrial products released by damaged cell can activate inflammation and act as signalling molecules that trigger production of pro-inflammatory cytokines (35). Mitochondrial ROS have been recently shown to be induced by inflammasome activators, and these mitochondrial ROS are in turn required for the activation of the inflammasome (36). Overall, important new molecular pathways involving mitochondrial damage and ROS production are being elucidated that profoundly affect not only DNA damage and activation of oncogenes, but also different aspects of inflammation, suggesting an important role as upstream regulators of cancer-related signalling pathways that promote inflammation and tumorigenesis (37).
Another example is represented by the aberrant expression of activation-induced cytidine deaminase (AID) that is induced in gastric epithelium by Helicobacter pylori (38). AID is a member of the cytidine-deaminase family that acts as a DNA- and RNA-editing enzyme. The H. pylori-mediated upregulation of AID resulted in the accumulation of nucleotide alterations in gastric cells, leading to the development of gastric cancer (38). In addition, the ectopic AID production, induced by TNF stimulation in human bile-duct cells, is associated with chronic biliary inflammation and the development of cholangiocarcinoma (39). Therefore, AID may link chronic inflammation to DNA damage in these tumours.
Key orchestrators at the intersection pathway of cancer-related inflammation
Among the molecular players involved in CRI, key endogenous promoters include transcription factors such as NFκB, signal transducer activator of transcription-3 (STAT3), and primary inflammatory cytokines such as IL-1β, IL-6, IL-23, and TNF-α (8,40-42).
NFκB is a key orchestrator of innate immunity and inflammation and it has recently emerged as potential molecular bridge between tumour cells and inflammatory cells (40). In both cellular contexts, NFκB operates downstream of the sensing of microbes or tissue damage by the toll-like receptor (TLR)-MyD88 pathway, the inflammatory cytokines TNF-α and IL-1β, and downstream of tissue damage that results in release of alarm signals. In addition, NFκB activation can be the result of cell-autonomous genetic alterations (amplification, mutations, or deletions) in cancer cells.
NFκB induces the expression of inflammatory cytokines, adhesion molecules, key enzymes in the prostaglandin synthase pathway (COX2), nitric oxide (NO) synthase, and angiogenic factors. In cancer and epithelial cells exposed to carcinogens, NFκB promotes cell survival and proliferation by the activation of genes encoding for proteins regulating cell cycle progression (e.g. cyclin D, c-myc) and apoptosis (e.g. cIAPs, A1/BFL1, Bcl2, c-Flip). Hepatocarcinogenesis significantly depends on NFκB activation in both parenchymal (hepatocytes) and non parenchymal cells of the liver (43). Strong genetic evidence, including tissue-specific gene targeting of components of the Ikk complex, such as IkappaB-kinase beta (IKKβ), has demonstrated a crucial role of NFκB in tumour promotion in prototypic tissues of CRI (e.g. gastrointestinal tract and liver). IKKbeta regulates gastric carcinogenesis via IL-1α expression, which is associated with anti-apoptotic signaling and cell proliferation (44). Accumulating evidence suggests that intersections and compensatory pathways may exist between the NFκB and HIF-1 systems (7,45,46) linking innate immunity to the hypoxic response.
The NFκB pathway is tightly controlled by inhibitors acting at different levels. Toll-interleukin receptor 8 (TIR8) is an example of a molecule that, by controlling inflammation, may protect against cancer. TIR8 also known as single immunoglobulin interleukin 1 receptor-related protein (SIGIRR), is a fringe member of the IL-1 receptor family with a single Ig domain, a long cytoplasmic tail and an altered TIR domain. It inhibits TLR and IL-1R signalling and is highly expressed in intestinal mucosa. TIR8 gene deficiency is associated with increased susceptibility to intestinal inflammation and carcinogenesis (47,48) as well as to B cell lymphoproliferation and autoimmunity (49,50). Another negative regulator of inflammation, namely A20 (also known as TNFAIP3), has been proposed to function as tumour suppressor in several human B-cell lymphoma (51).
Thus, balancing inhibitors and activators tunes the action of the NFκB pathway as an endogenous tumour promoter, acting in infiltrating leukocytes as well as in cells targeted by carcinogens. Evidence suggests that p50 homodimer-negative regulator is responsible for the sluggish NFκB activation in tumour-associated macrophages (TAM) and for their pro-tumour phenotype. Thus, NFκB appears to act as a rheostat tuned at different levels in florid inflammatory conditions predisposing to cancer (e.g. inflammatory bowel disease, IBD) and in TAMs sustaining the smouldering inflammatory milieu of established metastatic neoplasia (52,53). NFκB has also been recently involved in driving the M2 (alternatively activated macrophages) polarization of TAMs (54).
Along with NFκB, STAT3 is a point of convergence for numerous oncogenic signalling pathways (8). Constitutively activated STAT3 increases tumour cell proliferation, survival and invasion while suppressing anti-tumour immunity. The persistent activation of STAT3 also mediates tumour-promoting inflammation. STAT3 has this dual role in tumour inflammation and immunity by promoting pro-oncogenic inflammatory pathways, including NFκB and IL-6-gp130-JAK pathways and by opposing STAT1 and NFκB-mediated Th1 anti-tumour immune responses (55).
Soluble mediators of cancer-related inflammation
TNF-α plays a key role in the persistence of inflammation and established evidence demonstrates that tumour derived TNF-α sustains the growth and progression of syngenic, xenogenic and chemically induced tumours of skin, pancreas and bowels (31,56,57). In addition to cancer cells, other TNF-α producers have been identified in tumour microenvironment. Macrophages (58) and CD4+ cells (59) secrete TNF-α and trigger inflammation, as shown in a genetic model of liver cancer (43). TNF-α initiates a complex cross-talk between epithelial ovarian cancer cells and surrounding tissues, promoting the colonization of the peritoneum (59). Moreover, the constitutive production of TNF-α was associated with increased release of chemokines (CCL2, CXCL8, CXCL12), IL-6, VEGF and macrophage migration inhibitory factor (MIF-1). The mechanism of TNF-α action includes direct effects on tumour spread, via CXCR4; tumour cell survival, via CXCR4/CXCL12; but also stimulation of new blood vessels in the peritoneal tumour colonies, due to induction of CXCL12 and VEGF expression (60). A major source of inflammatory cytokines in the tumour microenvironment are tumour-associated macrophages (TAM) (61,62). TAMs promote wnt signalling through TNF in gastric cancer (63). These findings provide a rationale for the development of clinical protocols employing TNF antagonists in cancer therapy (64). Decoy receptor 3 (DcR3) is a member of the TNF receptor superfamily and has been involved in the control of MHC class II in TAMs (65). IL-1, IL-6, TNF-α, and the related receptor activator of NFκB ligand (RANKL) have long been known to augment the capacity to metastatize by affecting multiple steps in the dissemination and inflammation cascade (2,66,67). TAMs assist tumour behaviour in many ways, including producing cytokines, growth factors, and matrix-degrading enzymes (68-70). Kim et al. recently showed that tumour cell supernatants contained an inducer of cytokine production in macrophages. Unexpectedly, the tumour-derived macrophage activation was identified as a component of the extracellular matrix: versican, which is frequently up-regulated in human tumours. Versican, released by tissue damage, was found to be recognized by toll-like receptor 2 and 6 (TLR2/TLR6). Thus, in the Lewis lung carcinoma model this study identifies a cascade of amplification of metastasis initiated by tumour-derived versican acting on myeloid cells via TLR2/TLR6, leading to inflammatory cytokine production (71).
The involvement of IL-1 in tumour progression is well established. IL-1 expression is elevated in human breast, colon, lung, head and neck cancers, melanomas, and patients with IL-1 producing tumours have generally bad prognosis (72). IL-1 promotes tumour growth and metastasis by inducing several pro-metastatic genes such as metalloproteinases, chemokines, growth factors and TGF-β (73). IL-1 is also able to stimulate the expression of endothelial adhesion molecules such as ICAM-1 and VCAM-1 (74). Of particular importance are its effects on angiogenesis: IL-1 is a potent proangiogenic cytokine and VEGF release is also IL-1 dependent (75). In a pancreatic islet tumour model, a first wave of myc-driven angiogenesis is induced by the inflammatory cytokine IL-1 (76). IL-1α was also shown to play a pivotal role in the pathogenesis of liver cancer (77). On the other hand, IL-1α is possibly of importance in 3-methylcholantrene-induced fibrosarcoma, due to its efficiency in activating anti-tumour innate and specific immune responses, by acting as a focused adjuvant, through binding to IL-1R1 on immunesurveillance cells (78,79). Moreover, small amounts of IL-1α, which is homeostatically expressed in cells, but not secreted, can be poured out from necrotizing cells and serve as a ‘danger signal’ for mounting anti-tumour cell immunity (80). In 1993 the first study using IL-1R antagonist (IL-1Ra) treatment in mice transplanted with human melanoma cells demonstrated a strong reduction in the number of lung metastasis (81). Similar findings were reported in a model of hepatic metastasis: a single injection of IL-1Ra reduced tumor colonies by 50% and tumor volume by 70% (82). There is evidence of therapeutic efficacy in blocking the IL-1 pathway in humans with IL-1Ra and IL-1 inhibitors. In myeloma patients treated with dexamethasone and Anakinra (IL-1Ra) long progression-free intervals of over 3 years or 4 years were obtained in some patients (83) indicating a long stabilization of the disease. More potent IL-1 inhibitors are reaching clinical evaluation, in particular for autoimmune diseases, opening a possible future use in anti cancer therapies (84).
IL-6 is a key growth-promoting and anti-apoptotic inflammatory cytokine (85,86) and is also one of the effector signals of activated NFκB in the promotion of neoplasia. A clear pro-tumoral role for IL-6 has been demonstrated in multiple myeloma (87). Another example of IL-6-dependent tumour is the Hepatocellular carcinoma (HCC), the most common type of liver cancer. Naugler et al. (88) explored the molecular mechanisms to explain the higher incidence of liver cancer in male patients. It turned out that in females estrogens block IL-6 production in local immune cells (liver Kupffer cells). In IL-6 deficient mice or mice lacking the adaptor protein MyD88 (linked to NFκB signalling) the sex difference in susceptibility to cancer was absent (89). The importance of the autocrine IL-6 is confirmed also in other cancer types where it is demonstrated that IL-6 is an important activator of oncogenic STAT3 in lung adenocarcinomas and Jagged-1/Notch signalling in breast tumour mammospheres (90-92). New light was shed on the role of IL-6 in colitis-associated cancer (93,94). It was found that IL-6 produced by myeloid cells is a critical tumour promoter during intestinal carcinogenesis. IL-6 protects normal and pre-malignant intestinal epithelial cells from apoptosis and promotes the proliferation of tumour-initiating cells. These actions are mediated by gp130 and STAT3. Thus, the NFκB/IL-6/STAT3 cascade plays a key role in intestinal carcinogenesis. Interestingly, STAT3 also regulates the balance between IL-12 and IL-23 in the tumour microenvironment (95).
Another soluble component strictly associated with the recruitment of leukocytes in tumours is represented by chemokines (2,9,96,97). Recent results with gene-targeted mice have provided unequivocal evidence for a role of CC chemokines in carcinogenesis. Mice deficient in D6, a decoy and scavenger receptor for inflammatory CC chemokines, show increased susceptibility to skin carcinogenesis and colitis-associated cancer (98). The contribution of chemokines to angiogenesis and tumour promotion has been the object of intensive investigation. A variety of chemokines, including CCL2, CXCL12, CXCL8, CXCL1, CXCL13, CCL5, CCL17, and CCL22, have been detected in neoplastic tissues as products of either tumour cells or stromal elements (96,99). CXCL1 and related molecules (CXCL2, CXCL3, CXCL8, or IL-8) have an important role in melanoma progression by stimulating neoplastic growth, promoting inflammation, and inducing angiogenesis. Strong evidence demonstrates that levels of CCL2 are associated with TAM accumulation and that CCL2 may play an important role in the regulation of angiogenesis (1). Chemokines are also involved in the rapid turnover of myeloid-derived suppressor cells (100). Expression of chemokine receptors plays an important role in guiding metastasis. CXCR4 is the most frequently up-regulated chemokine receptor in cancer cells, and it is associated with advanced stages and metastasis (2). Recently, the chemokine receptor CX3CR1 has been reported to be up-regulated in human pancreatic cancer and to be involved in the perineural dissemination of this neoplasia along local nerve terminations (101).
Cellular components of cancer-related inflammation
TAM represent the major inflammatory component of the stroma of many tumours, able to affect different aspects of the neoplastic tissue (102,103). In several studies of human cancer, TAM accumulation has been associated with angiogenesis and with the production of angiogenic factors such as VEGF and platelet derived endothelial cell growth factor (1,104-109). TAMs accumulate in hypoxic regions of tumours, and hypoxia triggers a pro-angiogenic programme in these cells (104). A number of molecules with possible impact on angiogenesis have been shown to be expressed by macrophages in low-oxygen conditions, such as VEGF, TNF-α, basic fibroblast growth factor (bFGF), and CXCL8. Therefore, macrophages recruited in situ represent an indirect pathway of amplification of angiogenesis, in concert with angiogenic molecules directly produced by tumour cells (2,23). Under hypoxic conditions, tumour-secreted lactic acid promotes a IL-23/IL-17-mediated pro-inflammatory pathway in TAMs (110).
Myeloid cells in the tumour microenvironment are related to the angiogenic switch at different levels (111). VEGF and the related angiogenic factor placenta derived growth factor (PlDGF) have long been known to be potent monocyte attractants and to contribute to TAM recruitment (112). VEGF1R+ hematopoietic bone-marrow cells home to tumour-specific premetastatic sites. There, they form a recipient niche which favours secondary localization of cancer. Bv8, also known as prokinetic 2, modulates recruitment of angiogenic Gr+Mac1+ cells from bone-marrow (113) and promotes angiogenesis. Moreover, Gr+Mac1+ cells, presumably MDSC, have recently been shown to mediate resistance to anti-angiogenic therapy in various models (113). However, also mast cells, neutrophils and even effectors of the adaptive immunity (e.g. antibody-producing B cells activating macrophages) may sustain inflammatory reactions that promote cancer progression (114,115). In addition to producing angiogenic factors, such as chemokines and VEGF itself, myeloid cells are also a source of matrix-degrading enzymes (MMP) which mobilize VEGF from extracellular matrix stores. Recent results suggest that TAMs can promote tumour angiogenesis also via semaphorin 4D (116). Thus TAMs and related cells (MDSC, DC, polymorphonuclear cells (PMN)) represent an important indirect and alternative pathway of angiogenesis in the tumour microenvironment (117).
Concluding remarks
The link between inflammation and cancer is now well established, even in those cases where underlying chronic infections are absent. Two molecular pathways have been recognized and consist of an intrinsic pathway, driven by genetic alterations that cause neoplasia and activate the inflammation cascade, and an extrinsic pathway where inflammatory conditions facilitate cancer development. At the crossroads, the key orchestrators are represented by transcription factors (e.g. NFκB) that coordinate cytokine (e.g. TNF), chemokine and enzyme production. These factors recruit and activate leukocytes, mainly of the myelomonocytic lineage (112), producing and amplifying the inflammatory responses. Smouldering inflammation contributes to proliferation and survival of malignant cells, angiogenesis, metastasis, subversion of adaptive immunity and reduced response to chemotherapeutic agents.
CRI represents a target for pharmacological strategies that can modulate the host microenvironment. Primary pro-inflammatory cytokines, chemokines and their receptors represent prime targets of future biological therapies (57), and their clinical use is being experimented in cancer patients (118-120). Recently, new strategies are aimed at targeting TAM recruitment and polarization (53). For many years all efforts to treat cancer have concentrated only on the destruction/inhibition of cancer cells, CRI may represent a complementary perspective that can help the development of effective anti-tumour responses.
Acknowledgements
The authors are supported by the Italian Association for Cancer Research, Italian Ministry of Health, MIUR (Ministero dell’Istruzione Università e Ricerca).