Introduction
The human globin genes are organized into two separated clusters, alpha (α) located on chromosome 16 and beta (β), on chromosome 11. The structure of α- cluster is 5’-ζ-ψζ-ψα2-ψα1-α2-α1-θ-3’, with a regulatory element located upstream of the embryonic ζ-gene. For the β-cluster the order is 5’-ε-Gγ-Aγ-ψβ-δ-β-3’ with the locus control region located upstream of the ε-globin gene (1). A normal adult individual has approximately 97.5% hemoglobin (Hb) A, 2.0 % Hb A2 and 0.5 % Hb F (2).
Inherited Hb disorders with absent or reduced synthesis of particular globin chains are called thalassemias, while Hb variants or abnormal Hbs are structurally inherited changes of globin chains. Both Hb variants and thalassemias have been increasingly reported worldwide. To date, the HbVar database has noted more than 1,150 mutations (3). Although most of the Hb variants are non pathological variants, some could produce clinically relevant phenotypes when found in association with thalassemia or other hemoglobinopathies. Accurate diagnoses of these hemoglobinopathies are important for planning of appropriate management and genetic counseling. However, these are problematic in routine practice and usually require hematological, biochemical and molecular characterizations (4-6). Many α-thalassemias are caused by deletions of one (α+-thalassemia) or both α-globin genes in cis (α0-thalassemia). Although point mutations or small deletions and insertions have been reported for non-deletional α-thalassemia, their frequencies are relatively much lower (7). While interaction of α+-thalassemia with α0-thalassemia can cause Hb H disease, a thalassemia intermedia found commonly in the Southeast Asian populations and Chinese, homozygous α0-thalassemia leads to a fatal condition known as the Hb Bart’s hydrops fetalis. Most of the β-thalassemias are characterized by decreased or absent β-globin chain synthesis due to a variety of mutations. The majorities of the patients with homozygous β-thalassemia develop a severe form of anemia and are transfusion dependent from the first year of life (8).
Hb Grey Lynn is an α-globin variant due to a Leu to Phe substitution at a position 91 of α1-globin chain. It was once reported in individual of unknown ethnic origin from New Zealand (9) and once again as Hb Vientiane found in association with Hb E in a Laotian (3). We have now reported for the first time the interaction of Hb Grey Lynn (Vientiane) [α91(FG3)Leu>Phe (α1)] with a rare α+-thalassemia point mutation [α2 codon 36/37 (-C)] in adult Thai individual.
Materials and methods
Subjects
Ethical approval was obtained from the Institutional Review Board (IRB) of Khon Kaen University, Khon Kaen, Thailand (HE552035). The proband was adult Thai woman aged 28 years. She was discovered at the ongoing thalassemia screening program at provincial hospital. Because she had moderate anemia with Hb 90 g/L (10) with the presence of unknown Hb variant, her EDTA anticoagulated blood specimen was transferred to our center for further investigation.
Laboratory methods
Erythrocyte number, Hb level and erythrocyte indices were measured and calculated on blood cell counter (COULTER®HmX Hematology Analyzer; Beckman Coulter Co.-, Miami, FL, USA). Plasma ferritin concentration was assayed using chemiluminescent immunoassay on the Syncron LXi 725 Access® Clinical System (Beckman Coulter, USA). Hb analysis was done using cellulose acetate electrophoresis (CAE) (11), automated low performance liquid chromatography (LPLC) (Hb Gold; Drew Scientific Co., Ltd., Cumbria, UK), and capillary electrophoresis (Capillarys 2 Flex Piecing; Sebia, Lisses, France) (Figure 1), applying protocols provided by the manufacturers.
Genomic DNA was prepared from peripheral blood leukocyte as previously described (12). Testing for the presence of α0-thalassemia (--SEAand --THAI), α+-thalassemia (-α3.7 and -α4.2), Hb Constant Spring (αCSα or HBA2:c.427T>C) and Hb Pakse¢ (αPSα or HBA2:c.429A>T) commonly found in Thailand, was performed using polymerase chain reaction (PCR) based methods as described elsewhere (13,14). Previously described multiplex allele specific PCR assays were used to check the presence of mutations detected in patients previously referred to our laboratory: Hb Hekinan [α27(B8)Glu ® Asp or HBA2:c.84G>C], Hb Nakhon Ratchasima [α63(E12)Ala ® Val or HBA2:c.191C>T] and Hb Phnom Penh [α117(GH5)-Ile-α118(H1) or HBA1:p.Phe118_Thr119insIle] (15,16). DNA sequencing of amplified α1- and α2-globin genes were performed using specific primers and an ABI PRISMTM 3130 XL analyzer (Applied Biosystems, Foster City, CA, USA) (17).
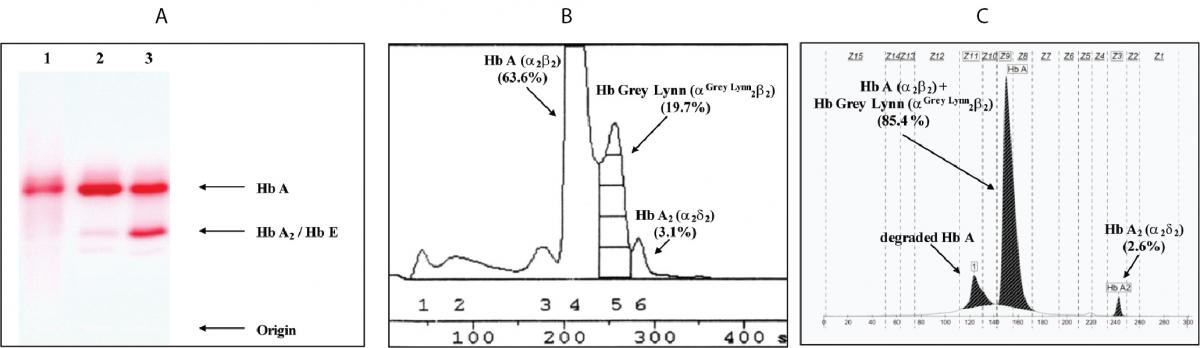
Figure 1. Hb analysis using cellulose acetate electrophoresis (A). 1-3 are the proband, normal control and Hb E carrier, respectively. B and C are Hb analysis profiles of the proband using automated LPLC and capillary electrophoresis, respectively.
Identification of Hb Grey Lynn mutation
The PCR-RFLP assay was developed for identification of Hb Grey Lynn mutation on α1-globin gene. Selective amplification of α1-globin specific fragment (975 bp) was performed by PCR using primers C1 (5¢-TGG AGG GTG GAG ACG TCC TG-3’) and B (5’-GAG GCC CAA GGG GCA AGA AGC AT-3’) (16). The PCR product was digested to completion with HindIII restriction enzyme (5’-A▼AGCTT-3’) (New England Biolabs, Beverly, MA, USA) (NEBcutter V2.0, http://tools.neb.com/NEBcutter2/). After digestion, normal control fragment is digested into 627 bp and 348 bp fragments whereas the 975 bp fragment is specific for Hb Grey Lynn.
Identification of α+-thalassemia [α2 codon 36/37 (-C)]
An allele-specific PCR assay was developed for identification of the α2 codon 36/37 (-C) α+-thalassemia mutation. In this assay, primer αG58 (5’-AGG ATG TTC CTG TCC TTC CCA-3’), specific to this mutation is used with a common primer C3 (5’-CCA TTG TTG GCA CAT TCC GG-3’) to produce the 634 bp mutation specific fragment. The 391 bp fragment produced with primers αG17 (5’-AGA TGG CGC CTT CCT CTC AGG-3’) and C3 is used as an internal control of PCR amplification. The PCR reaction mixture (50 µl) contains 50-100 ng of genomic DNA, 30 pmol of primers αG58 and C3, 3 pmol of primer αG17, 200 µM dNTPs, 0.75 M betaine, 5% DMSO and 1.5 unit Taq DNA polymerase (New England Biolab, Inc., USA) in 12 mM Tris-HCl buffer pH 8.3, 50 mM KCl, 1.5 mM MgCl2, 0.01% gelatin. PCR amplification was carried out on a T-Personal Thermocycler (Biometra; GmbH, Gottingen, Germany). After initial heat activation step of 3 minutes at 94°C, 30 cycles were repeated under the following conditions: 30 sec at 94 °C, 30 sec at 62 °C and 2 min at 68 °C. The amplified product was separated on 1.5 % agarose gel electrophoresis and visualized under UV light after ethidium bromide staining.
Results
Hematologic analysis of the proband revealed Hb 90 g/L and MCH 26.3 pg (Table 1). Plasma ferritin concentration (75 µg/L) was within reference range (20-300 µg/L) (18) which ruled out iron deficiency anemia. Other hemolysis markers including reticulocyte count, serum bilirubin, haptoglobin and hemopexin levels were not examined at that time. Although initial Hb analysis on cellulose acetate electrophoresis identified no abnormal Hb (Figure 1A), analysis using LPLC analyzer identified 63.6 % Hb A, 3.1 % Hb A2 and 19.7 % unknown Hb separating between Hb A and Hb A2 at the Hb E-window, quite resembling Hb E heterozygote (Figure 1B). Inconsistency with this finding included a negative result for the dichlorophenolindophenol (DCIP) screening test for Hb E and the result of Hb analysis using capillary electrophoresis, which showed 85.4 % Hb A and 2.6 % Hb A2, with no Hb E nor abnormal Hb peak noted (Figure 1C). Routine molecular screening for α-thalassemia mutations commonly found in Thailand revealed negative results. Screening for Hb Hekinan, Hb Nakhon Ratchasima and Hb Phnom Penh mutations revealed negative results. Further DNA sequencing of α1 and α2-globin genes of the patient identified the CTT (Leu) to TTT (Phe) substitution at codon 91 on α1-globin gene corresponding to the Hb Grey Lynn (9) or Hb Vientiane (3) (Figure 2A) (Hb Grey Lynn and Hb Vientiane are synonyms) and identified a (-C) deletion between codons 36 and 37 of α2-globin gene [α2 codon 36/37 (-C)] (or HBA2:c.112delC) leading to a premature termination at a new codon 48 as shown in Figure 2B. The patient was therefore confirmed to be a compound heterozygote for Hb Grey Lynn and α+-thalassemia [α2 codon 36/37 (-C)], a hitherto un-described condition.
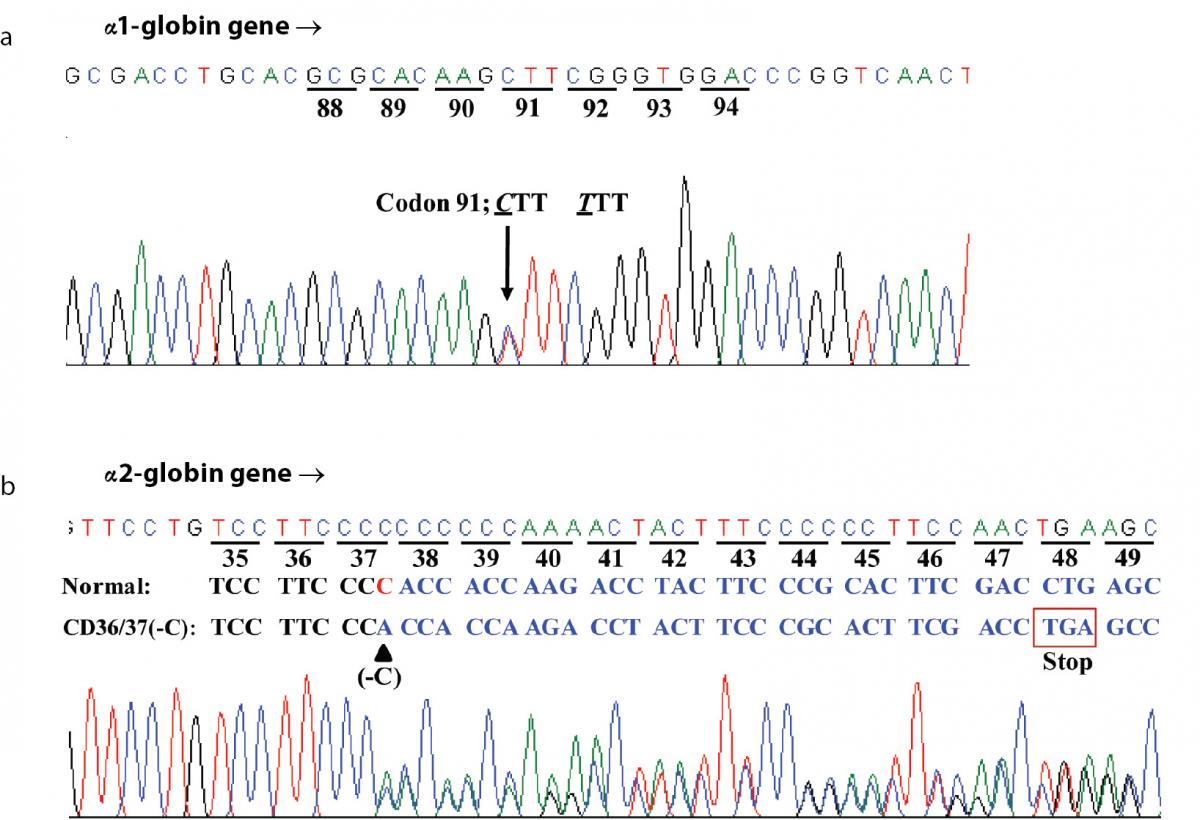
Figure 2. DNA sequence profiles of the proband demonstrating a CTT (Leu) to TTT (Phe) mutation at codon 91 of α1-globin gene corresponding to the Hb Grey Lynn (A) and a C deletion between codons 36 and 37 of α2-globin gene leading to a premature termination at a novel codon 48 and α+-thalassemia (B).
As the Hb Grey Lynn mutation eliminates a HindIII restriction site at codon 91 of α1-globin gene, PCR-RFLP method, as shown in Figure 3A, was confirmed as applicable for detection of this variant. Regarding targeted detection C deletion between codons 36/37 of α2-globin gene (or HBA2:c.112delC), reliable results were achieved by using allele specific PCR assay as shown in Figure 3B. Hematological parameters of this Thai patient were compared with those of the previously documented cases of compound heterozygote for Hb Grey Lynn / α0-thalassemia (9), double heterozygote for Hb Vientiane / Hb E (3) and heterozygous Hb Treviso with the same mutation on α2-globin gene [α91(FG3)Leu®Phe (α2)] recently reported (19) (Table 1). As compared to those observed in a compound heterozygote for Hb Grey Lynn / α0-thalassemia, relatively normal MCV and MCH values were noted in a compound heterozygote for Hb Grey Lynn / α+-thalassemia (the proband), double heterozygote for Hb Grey Lynn / Hb E and Hb Treviso trait.
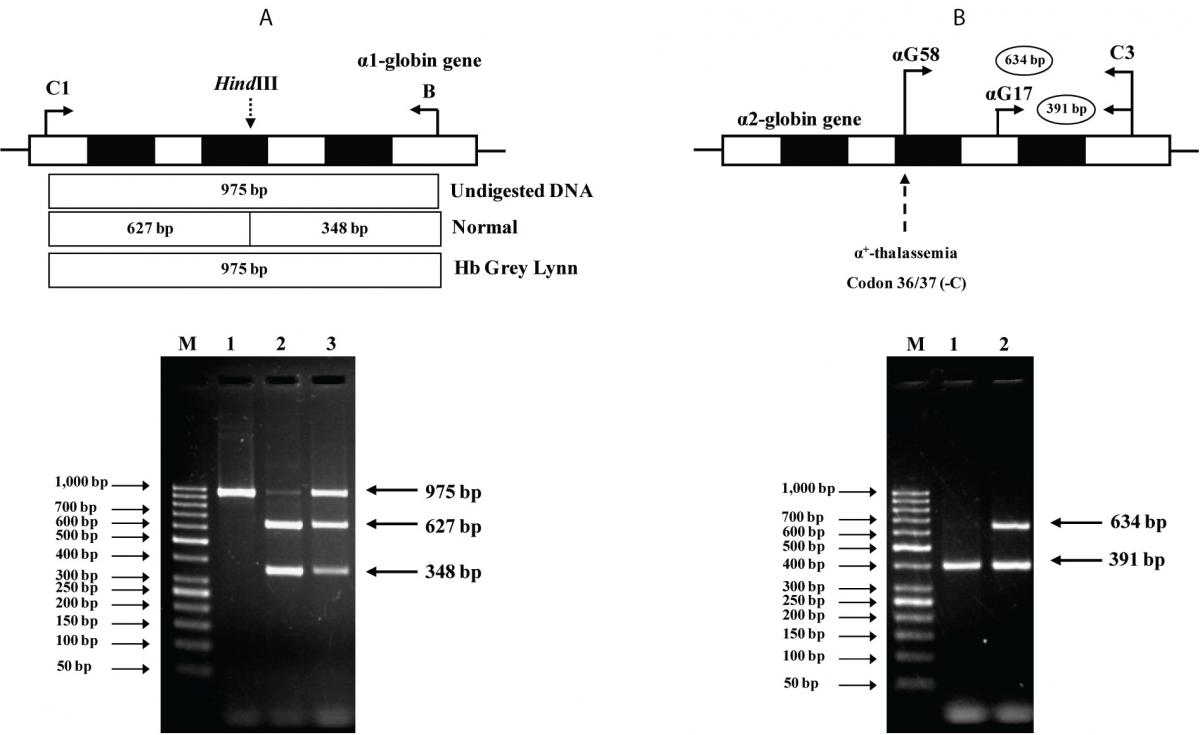
Figure 3. A: Identification of the Hb Grey Lynn mutation by PCR-RFLP assay using HindIII digestion. The locations of primers C1 and B used to generate an α1 specific fragment of 975 bp by PCR are shown. Normal control fragment is digested into two fragments with 627 bp and 348 bp in lengths while the undigested 975 bp fragment indicates the presence of Hb Grey Lynn mutation. M represents the GeneRuler 50 bp Plus DNA ladder. 1: amplified DNA of the proband, 2: HindIII-digested amplified DNA of a normal control, and 3: HindIII-digested amplified DNA of the proband with Hb Grey Lynn.
B: Identification of α+-thalassemia [α2 codon 36/37 (-C)] by allele specific PCR. The locations and orientations of primers (αG17 & C3) and (αG58 & C3) used respectively to produce the 634 bp α2 codon 36/37 (-C) mutation specific fragment and the 391 bp internal control fragment are depicted. M represents the GeneRuler 50 bp Plus DNA ladder. The α2 codon 36/37 (-C) mutation was identified in the proband (lane 2) but not in a normal control (lane 1).
Discussion
We report for the first time in this study interaction of Hb Grey Lynn (Vientiane) [α91(FG3)Leu>Phe (α1)] with a rare α+-thalassemia point mutation [α2 codon 36/37 (-C)]. Hb Grey Lynn has been documented in individual of unknown ethnic background in Auckland, New Zealand in the presence of α0-thalassemia (9). It has been noted again in 2007, as Hb Vientiane in association with Hb E in a Laotian subject (3). In both patients neither Hb H nor thalassemia phenotype was observed. The variant exhibited mild instability and altered O2 affinity since substitution of Phe for Leu at position 91 of α1-globin chain could affect heme interactions and α1β2 contact (9). Similar findings have been noted for another Hb variant with the same mutation but described alternatively on the α2-globin gene i.e. the Hb Treviso [α91(FG3)Leu-Phe(α2)] found in an Italian subject (19).
As shown in Table 1, it seems likely that the mild instability and decreased O2 affinity of these Hb variants do not produce significant hematological phenotype of the patients. However, with the lack of additional laboratory results especially for measurement of hemolysis markers e.g. reticulocyte count, serum bilirubin, lactate dehydrogenase, and haptoglobin, we could not exclude in vivo hemolysis. This is a limitation of our case report although no evidence was observed in the cases reported previously (9,19). The moderate normochromic normocytic anemia observed in the Thai patient described here could be due to the interaction of Hb Grey Lynn with α+-thalassemia. Iron deficiency has been excluded. In fact, it has been shown previously that thalassemia and hemoglobinopathies rather than iron deficiency are major causes of anemia in the region (20,21).
Table 1. Hematological parameters of the proband with Hb Grey Lynn and α+-thalassemia as compared to those of subjects with Hb Grey Lynn and α0-thalassemia, Hb Vientiane / Hb E and Hb Treviso trait, reported previously.
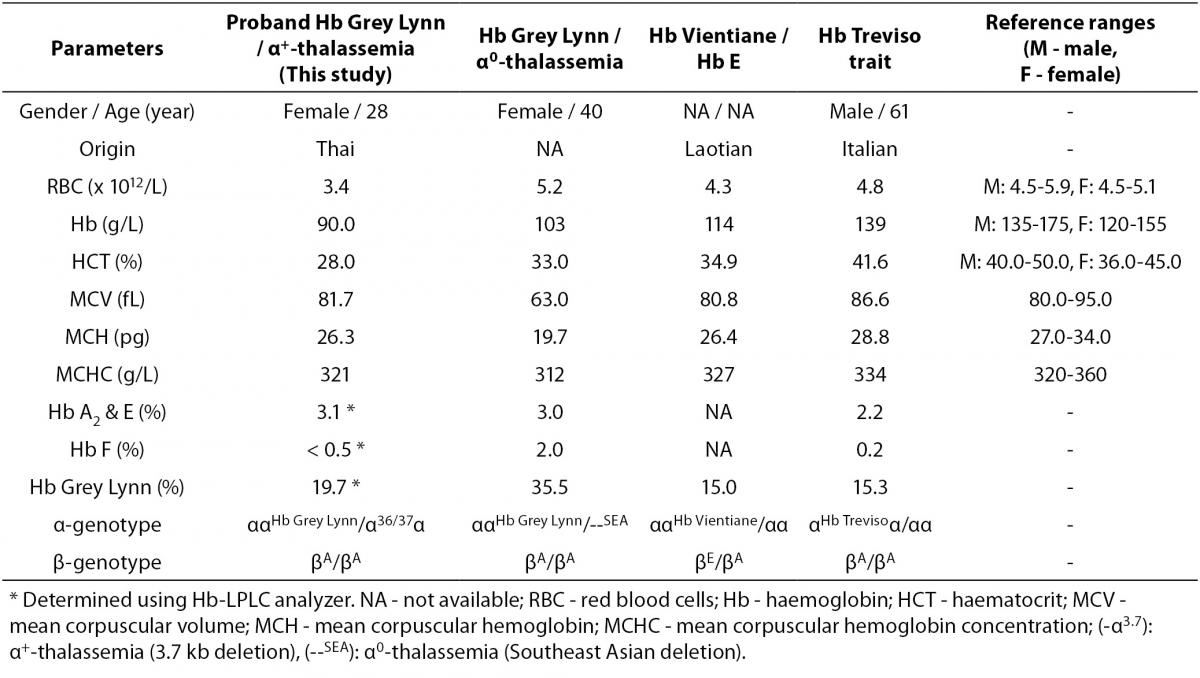
Among approximately 400 α-globin chain variants documented, about 20 variants have been found on both α1- and α2-globin genes (3). Although these mutations lead to identical α-globin chain variants, they are considered as different mutational events. The average proportion of Hb variant due to α2-globin gene mutation is usually higher than that of the same mutation on α1-globin gene for the higher expression of α2-globin gene (22). This however could be changed with a genetic compound heterozygosis. As shown in Table 1, in heterozygote form the level of Hb Grey Lynn is detected at around 15 % (Hb Treviso trait). However, elevated levels could be seen with co-inheritance of α+-thalassemia (the proband; 19.7%) and α0-thalassemia (35.5%), likely because of the decreased availability of normal α-globin chain with these conditions.
Most of the α-thalassemias in Thai and other Southeast Asian populations are caused by gene deletions (23). The finding of a relatively rare and previously un-described in Thai population, the α+-thalassemia due to codon 36/37 (-C) mutation indicates that α-thalassemia due to point mutation may not be uncommon in the population. This frameshift mutation results in a premature termination of α-globin chain synthesis at codon 48 which should trigger an increased mRNA degradation via a nonsense-mediated mRNA decay (NMD) mechanism (24). According to currently available literature, this α-thalassemia associated mutation has not been described before, though it was deposited as an α2 codon 37 (-C) at the HbVar database (3). No ethnic background and clinical phenotype of the case is provided. Due to the lack of family analysis of our patient who was a compound heterozygote for this unpublished α+-thalassemia mutation on α2-globin gene and Hb Grey Lynn on α1-globin gene, we do not know for certain if both mutations are located in cis or in trans on the α-globin gene cluster. Identification of Hb Grey Lynn but not this α+-thalassemia mutation in two cases reportedly previously (Table 1) indicates likely that these two mutations are located intrans. We do not know the exact prevalence of this non-deletional α+-thalassemia mutation in Thai population. Nonetheless, it is conceivable that interaction of this α+-thalassemia mutation with α0-thalassemia and/or Hb E could lead to a severe Hb H or AEBart’s disease commonly encountered in the region (25). Accurate diagnosis is essential for genetic counseling and treatment planning of the case.
It is noteworthy as shown in Figure 1 that diagnosis of Hb Grey Lynn at routine Hb analysis might be problematic as it co-migrates with Hb A on cellulose acetate electrophoresis and is not completely separated from Hb A on chromatography. In addition, a substitution of Phe for Leu at position 91 of α1-globin chain does not modify the net charge of the Hb molecule, capillary electrophoresis cannot detect it either. This result highlights the importance of timely diagnosis of abnormal Hbs by multiple methods at hematological, Hb analysis and molecular levels. Combined analyses using cellulose acetate electrophoresis, LPLC or HPLC and capillary electrophoresis would be a better alternative when possible. The result of these Hb analyses should provide useful information for selection of appropriate molecular testing in the final diagnosis of such variants. The characterization methods described here should prove useful on this matter.
Acknowledgements
This work was supported by grants from the National Research University (NRU) program of Khon Kaen University and the Office of the Higher Education Commission, Ministry of Education. Kritsada Singha is supported by the Royal Golden Jubilee PhD program of the Thailand Research Fund (TRF) and the Centre for Research and Development of Medical Diagnostic Laboratories (CMDL), Faculty of Associated Medical Sciences, Khon Kaen University, Thailand. We thank Ian Thomas for editing the manuscript.