Introduction
Parkinson’s disease (PD) is etiologically, genetically and pathologically complex and heterogeneous disease (1). It is estimated that most cases of PD (>95%) are sporadic and have late age onset, yet small but growing subset of individuals has a single gene defect as the cause of the disease (2,3). In the last group, at least ten loci associated with PD have been identified so far: PARK1, PARK2, PARK3, PARK4, PARK5, PARK6, PARK7, PARK8, PARK9 and GBA (4).
Genome-wide study of multiplex PD family offered evidence about connection of those loci which are on the different chromosomes (5-7). Genetically defined familial forms of PD offer insights into molecular signaling pathways that modulate protein degradation and mitochondrial homeostasis sustaining the selective neurodegenerative process in typical Parkinsonism (8). Genetic heterogeneity of monogenic form of PD points to the great pathophysiological complexity of multifactorial forms of PD (4). Linkage analysis and gene expression studies indicate a very large number of candidate genes for PD (5-9). Also, variation of genes which are associated with sporadic cases of PD can increase or decrease risk for the disease. Moreover, the concept of susceptibility genes allows the involvement of gene-gene and gene-environment interaction in sporadic PD (10). Previous studies of genes identified as candidate genes for PD indicate that there are many interactions between mutated genes.
The up regulated genes are clustered in cell adhesion/cytoskeleton, extra cellular matrix components, cell cycle, protein modification/phosphorylation, protein metabolism and transcription, and inflammation/hypoxia (e.g., key iron and oxygen sensor EGLN1) (11). Although, most interactions between their protein products cause cell death which is common incidence in pathophysiology of PD, oxidative stress is considered as primary cause of dopamine neurons death (4,12). In recent times, cell adhesion is often brought into relation with PD (13,14). Cell adhesion molecules play a central role in neural development and are also critically involved in axon regeneration and plasticity of synapses in adult nerve system (13,15). It is estimated that, L1 adhesion molecule was capable of stimulating survival and differentiation in the mid-brain neurons which degenerate in Parkinson’s disease (13). The neural cell adhesion molecule (NCAM) participates in adhesion and neurons outgrowth during nervous system development. In the adult brain, NCAM is considered to be involved in neuronal sprouting and synaptic remodeling (15). The increased levels of cytokine levels described in PD could induce the expression of vascular adhesion molecules (VCAM-1-VLA-4) and intracellular adhesion molecules (ICAM-1-LFA-1) (16). Identification of mutations in genes that lead the development of PD with high penetrance, show that the disease can have a significant genetic component. In terms of discovering new information from the literature, especially for candidate genes for various diseases, Peterlin and Hristovski have developed biomedical support discovery system (BITOLA) (17). By using this system were identified candidate genes of interest for multiple sclerosis (MS) and bilateral polymicrogyria (BPP) (18-20). Methods of integration data from the literature, using BITOLA tools with existing genomic and transcriptomics data can reveal new potential candidate genes for Parkinson’s disease.
The main goal of this research was to identify candidate genes for PD which correspond to following criteria nominated by authors in design of study: to show a specific pattern of expression in brain tissue, to be involved in processes of cell adhesion and cell death, and that so far in the literature are not brought into relation with PD.
Materials and methods
BITOLA system search strategy
For the detection of functional relation between potential candidate genes and pathogenetic mechanisms associated with PD a strategy of integration results from transcriptomics studies with approach of discovering new candidate genes from bibliographic data bases using BITOLA system was designed (17). Information about chromosome loci, tissue-specific expression and the function of potential candidate genes and their association with some genetics disorders were extracted from databases: Medline (21), Locus Link (22), Gene Cards (23) and Online Mendelian Inheritance in Man (OMIM) (24). In order to include some gene as candidate genes in the list of candidate genes for PD, that gene had to meet following criteria nominated by authors: to show a specific pattern of expression in brain tissue, to be involved in processes of cell adhesion and cell death, and that so far in the literature have not been brought into relation with PD. To find new potential biomedical relation between PD and pathogenetic mechanisms (cell adhesion and cell death) the BITOLA system was used (17).
The BITOLA system is an interactive literature-based biomedical discovery support system. The purpose of the system is to help biomedical researchers make new discoveries by revealing potentially new relations between biomedical concepts. The set of concepts currently contains Medical Subject Headings (MeSH), which is used to index Medline, and human genes from The Human Genome Organization (HUGO). The potential new relations are discovered by mining the Medline database (21).
The system is available in two versions: ‘’closed discovery’’ and ‘’opened discovery’’. Closed discovery allows the input of a single concept and generates potential explanations of the relationship between two entities. It does by searching published literature to finds intermediate links. Open discovery allows the input of a single concept, then categories for first-order relatives of that concept, then categories for relatives of those first order concepts. The BITOLA system was used according to the authors proposed instruction (17). Discovery algorithm for discovering new relations between medical concepts is described in Table 1 (18).
Table 1. The algorithm for discovering new relations between medical concepts (18)

Discovery algorithm for finding new relations between the given concepts was adapted to PD. As the concept X we nominated Parkinson’s disease, then concept Y is cell function and concept Z is gene or gene products. The main goal was to first find all the concepts Y (cell function) related to the starting concepts X (PD). Then all the concepts Z (gene or gene product) are found. As the last step, we check if X (PD) and Z (gene or gene product) appears together in the medical literature, then we evaluated the proposed (X (PD), Z (gene or gene product)) pairs and select among them those that deserve further investigation. If the chromosomal region of PD matches the location of the related genes (Z) and if there are no MEDLINE documents mentioning both the PD and the genes Z, then the genes Z can be proposed as candidate genes for X (PD). Our discovery algorithm we integrated in opened BITOLA system and all steps using BITOLA described in Figure 1. As beginning concepts X was entered the name Parkinson’s disease. After limiting the related concepts Y by the semantic type Cell function, 53 concepts were obtained corresponding to MeSH descriptions. According to the research strategy, the cell adhesion and cell death were chosen as the most suited to PD. Using those concepts, all related concepts Z of the semantic type gene or gene products were searched and further limited to those matching chromosome location and discoveries only. The all chromosome loci which in literature associated with PD were examined.
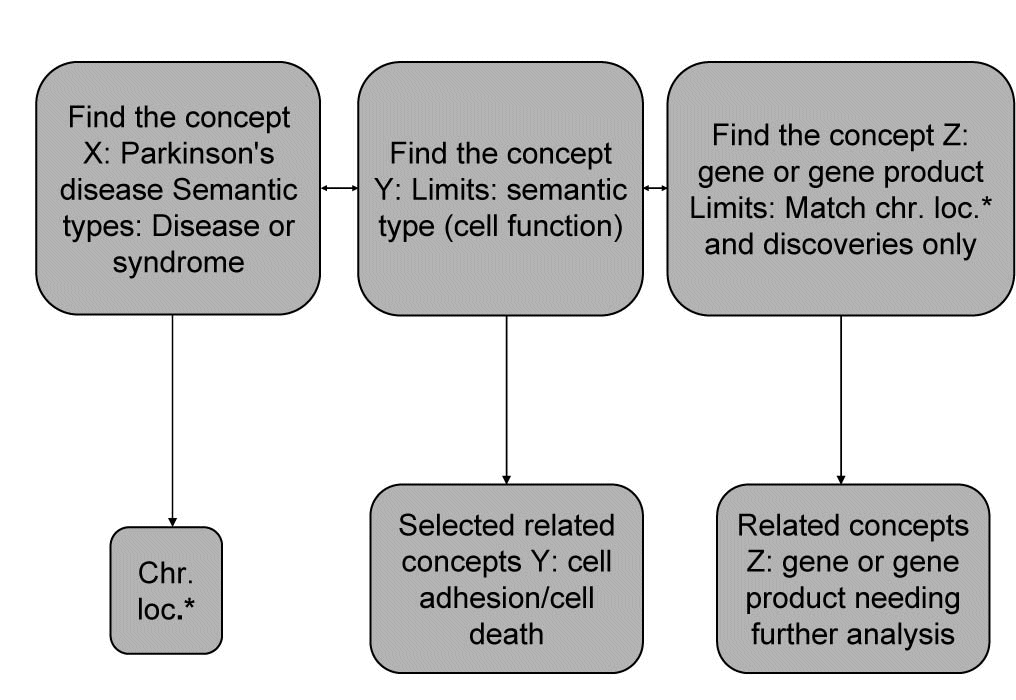
Figure 1. Discovery algorithm adapted to PD and integrated into BITOLA system. *Chr.loc. – chromosome loci.
In a closed BITOLA system the occurrence of the genes from list of candidate genes for PD together with PD in the literature were tested. For each gene from the list of candidate genes for PD the same discovery algorithm was integrated in closed BITOLA system. The concept X was Parkinson’s disease and the concept Z was gene from the list of candidate genes for PD.
Identification of transcriptomics studies
For identification of transcriptomics studies electronic data base Medline was researched (21). In this research were included all studies until December 2007. Terms used to search databases are MeSH terms and text words in English: “Transcriptome analysis” AND “Parkinson’s disease”, “Microarray analysis” AND “Parkinson’s disease” AND “Gene”. The above terms are used in combination with the terms “genetic”, “genomic association” and “study”. The results are set under Limits in the ‘’English language’’. Also, all references of identified studies were examined. The studies without full text were excluded from research.
Results
Using the adapted discovery algorithm to the PD and its integration into the opened BITOLA system, we searched all the related concepts Z (gene or gene product) and further limited them to those matching the chromosome loci described in Table 2. In this manner 181 genes were suggested by the opened BITOLA system.
Table 2. The chromosome regions examined in opened BITOLA system
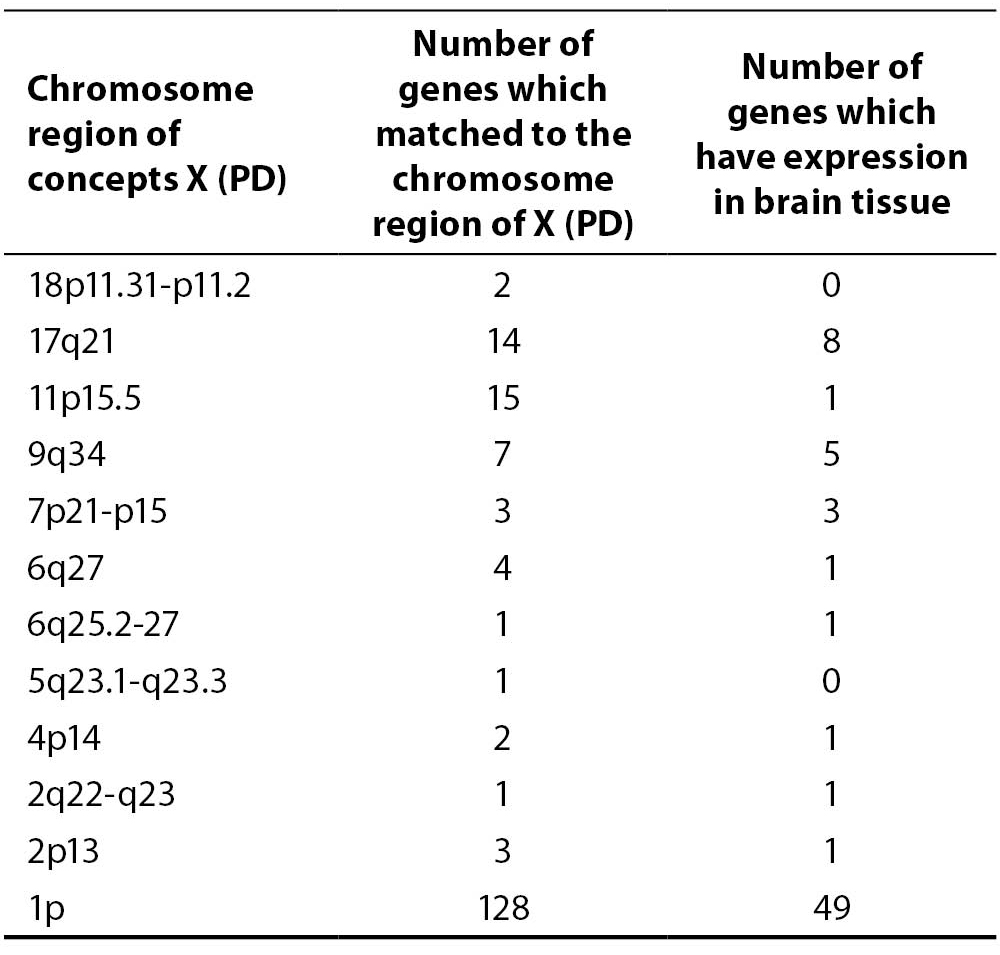
By further analysis of those genes we have excluded 110 genes whose expression pattern did not preferentially included the brain tissue which is target in the PD. According to the information from Locus Link (22), Gene Cards (23) and OMIM (24) about tissue-specific pattern and the cell function (cell adhesion and cell death) we have included 17 genes of the remaining 71 genes as potential candidate genes for PD (Table 3).
Table 3. The genes extracted from the opened BITOLA system for the further analysis
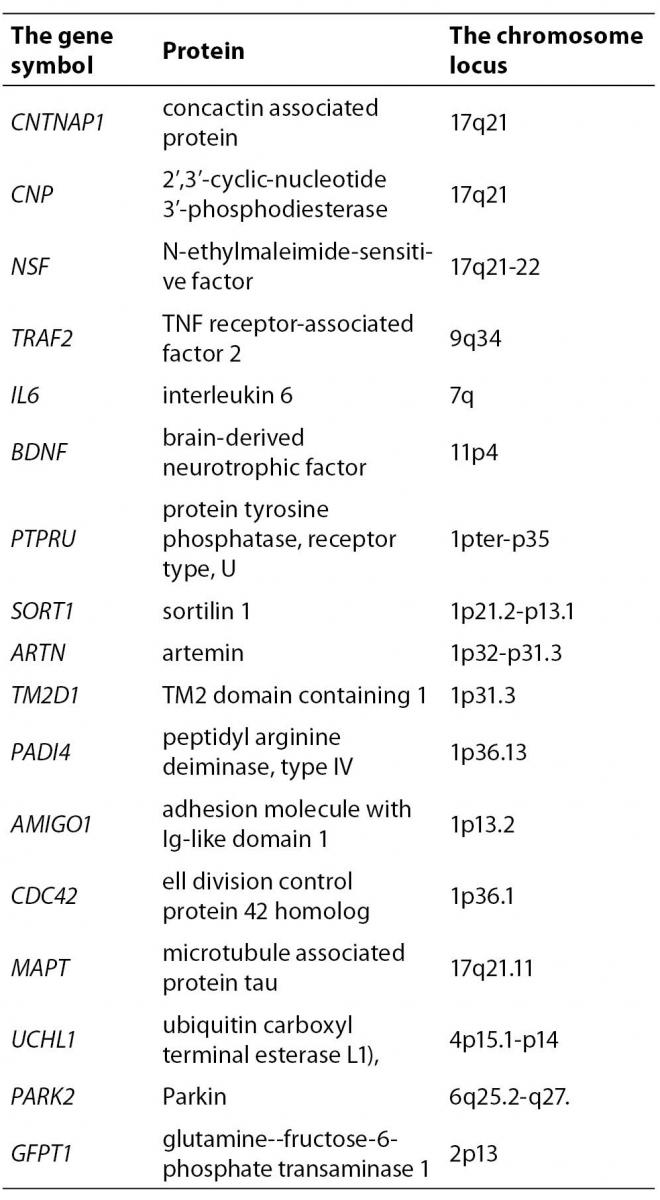
In the next step, in a closed BITOLA system we have tested the occurrence of those genes together with PD in the literature. The frequencies of X (PD) and Z (CNTNAP1, NSF, CNP, TRAF2, IL6, BDNF, PTPRU, SORT1, ARTN, TM2D1, GFPT1, PADI1, CDC42, MAPT, UCHL1, PARK2 and AMIGO1) in Medline documents is shown in figure 2.
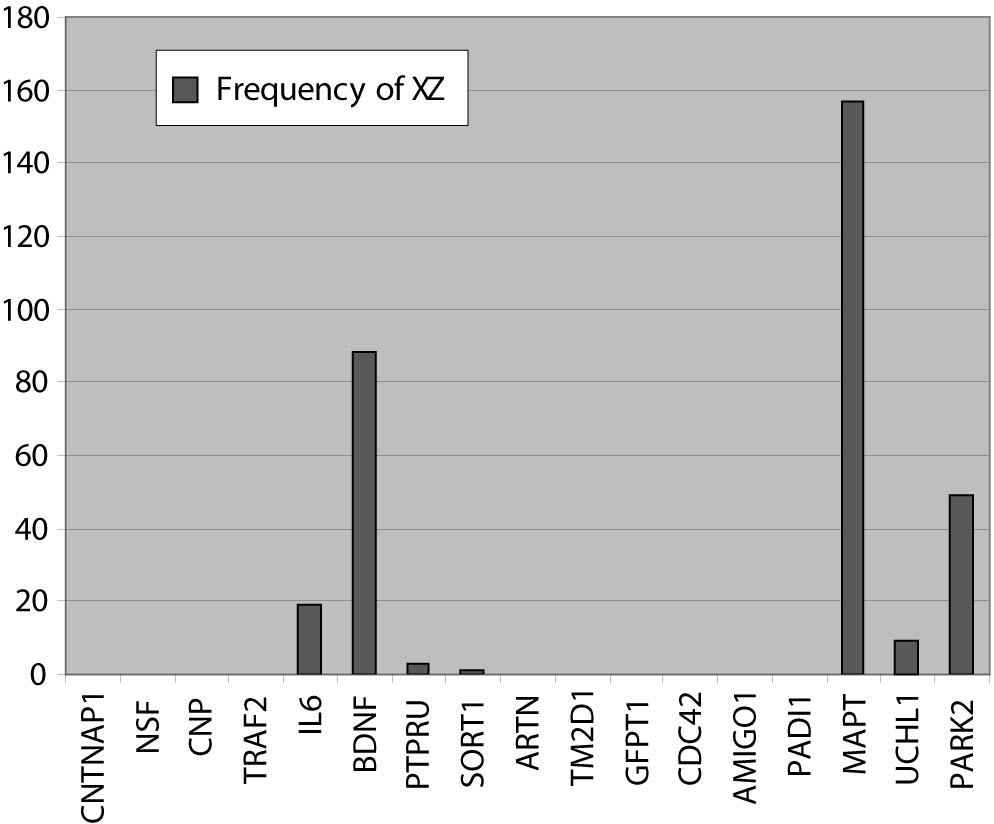
Figure 2. The frequencies of X (PD) and Z (gene or gene product) in Medline records.
IL6, BDNF, MAPT and PARK2 genes had the highest frequencies. The gene IL6 appears with PD in 19 Medline records, BDNF and PD in 88 records, MAPT and PD in 157 records and PARK2 and PD in 49 Medline records. This could be explained by the facts that those genes were researched in population of patients with PD and the role of the genes MAPT and PARK2 in pathogenesis of PD is confirmed. The PTPRUgene with concept X (PD) appears in 3 Medline documents. However, none of the listed studies do confirm its role in PD, so we didn’t exclude this gene. The genes CNTNAP1, CNP, TRAF2, SORT1, ARTN, TM2D1, NSF, PADI4, CDC42, GFPT1 and AMIGO1 do not appear together with the concept X (PD) in Medline documents.
One of the rules of discovery algorithm is that association between concept X (PD) and the concept Z (gene or gene product) must not exist. So we exclude from further analysis genes IL6, BDNF, MAPT and PARK2. However, their existence in list of offered genes in the BITOLA system is important because it is implicates on potential role of the BITOLA system in (re)identification disease candidate genes. According to the tissue-specific pattern of the remaining twelve genes and the facts that those genes are not researched into relation with PD, they could be proposed as interesting candidate genes for further analysis.
Searching the Medline database by “Transcriptome analysis” AND “Parkinson’s disease”, “Microarray analysis” AND “Parkinson’s disease”, “Gene expression profiling” AND “Parkinson’s disease” 41 documents were identified. The six Medline records corresponded to the criteria which were set up in design of study. After analysis of all available studies (26-31), the genes that the determined study offered as possible candidate genes for PD were selected.
In the next step, were compared and integrated results of analyzing transcriptomics studies and the list of offered genes in the opened BITOLA system. The genes NSF, GFPT1, MAPT, UCHL1, PARK2 and CDC42 found that overlap in those two groups of results.
Discussion
By integration of results obtained by using bioinformatics tool BITOLA and analyses of transcriptomics studies, we compiled the list of potential candidate genes for PD. The twelve genes are chosen as potential candidate genes (CNTNAP1, NSF, CNP, TRAF2, PADI4, PTPRU, SORT1, ARTN, TM2D1, CDC42, GFPT1 and AMIGO1) whose role in PD should be further explored. The most interesting of those twelve genes are NSF, CDC42 and GFPT1 because they appear in our BITOLA results and results of analyzing of transcriptomics studies.
N-ethylmaleimide sensitive factor (NSF) is an ATPases associated with various cellular activities protein (AAA), broadly required for intracellular membrane fusion (32,34). It does seem to interact with other proteins, such as the AMPA receptor subunit, GluR2, and beta2-AR and is thought to affect their trafficking patterns. Recently, it has been shown that NSF can be regulated by hydrogen peroxide. H2O2 is thought to inactivate NSF through oxidation of the Cys264 in NSF-D1 (32). Consistently, mutation of Cys264 to threonine eliminates the sensitivity of NSF to H2O2 (33). While this might suggest that NSF could be a redox sensor in the cell, whose activity is decreased when the oxidation state of the cytosol increases (32).
Interestingly, NSF gene is located nearby MAPT gene. Mutations in the tau gene, MAPT, causes familial frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17), and common variation in MAPT is strongly associated with the risk of progressive supranuclear palsy (PSP), corticobasal degeneration and, to a lesser extent, AD and PD, implicating the involvement of tau in common neurodegenerative pathway(s) (35). The genomic complexity around the MAPT locus is emphasized not only by complex arrangements of duplications close to the NSF gene, but also in a recently identified de novo micro deletion of 500–600 kb of the locus in individuals with developmental delay and learning disabilities (35). On the basis of previous facts, interaction between MAPT and NSF genes seems to be an interesting researching potential.
Neuronal apoptosis or programmed cell death (PCD) is a crucial process occurring not only during normal development and tissue turnover, but also in pathological situations such as stroke and neurodegenerative diseases (36). Neuronal PCD involves the activation of a number of enzymes and genes and is regulated by specific growth factors, such as neurotrophins, which promote survival of particular neuronal populations by binding to specific cell surface receptors. Over expression of activated Rac1 or Cdc42 in SCG neurons maintained in the presence of NGF induced apoptosis, whereas expression of dominant negative mutants of Cdc42 or Rac1 blocked apoptosis following NGF withdrawal. Furthermore, Cdc42-induced death was prevented by co expressing the c-Jun dominant negative FLAGΔ169 (37). Taking into account the fact that CDC42 is a key component of the cell death machinery in sympathetic neurons (37), its potential the role in PD should be further considered.
Glutamine-fructose-6-phosphate transaminase 1 (GFPAT1) is the rate-limiting enzyme of the hexosamine pathway that has been implicated in the pathogenesis of diabetic nephropathy (38). Glucosamine 6-phosphate is subsequently converted to uridine diphosphate N-acetylglucosamine, which is used for the O-glycosylation of intracellular proteins. Although this gene is associated with diabetic nephropathy, it is differentially regulated in PD and may play a role in sporadic cases of PD and role in sporadic PD and represent candidate for as yet unidentified disease-causing genes (31).
Taking into account the fact that genes of NSF, CDC42 and GFPT1 have not been brought into correlation with the PD, as opposed to gene PARK2, MAPT and UCHL1, and that they occur simultaneously in two groups of results, their significance for the PD may represent a potential target for further research. On the basis of above mentioned, the role of NSF, CDC42 and GFPT1 genes, as well as the role of 11 genes that are selected in the list of candidate genes for PD in opened BITOLA system, which do not overlap with the results of the analysis of transcriptomic studies could be readily tested by mutation screening of PD patients.
By this approach the new genes previously not known to be involved in the disease but transcriptionally co regulated with or physically interacting with members of these pathways could be identified. Any single functional genomics measure may suffer from incomplete coverage, imperfect sensitivity, or low specificity, but the combination of data from different studies and obtained by different methods can highlight candidate genes with increased confidence.